
January 24, 2018
Gregg Shapiro, Esq.
Chief of the Affirmative Civil Enforcement Unit
United States Attorney’s Office
District of Massachusetts
1 Courthouse Way, Suite 9200
Boston, MA 02210
Re. How Forest Misled the FDA, DOJ, USAO, and the Public about the Results of Celexa Study MD-18
Dear Mr. Shapiro:
On September 15, 2010, Forest Laboratories, Inc. and Forest Pharmaceuticals, Inc. (“Forest”) entered into a series of agreements with the United States Attorney’s Office for the District of Massachusetts (“USAO”).
First, Forest agreed to plead guilty to one count of obstruction and two counts of distributing a misbranded drug under the Food, Drug, and Cosmetic Act. The third count specifically related to Forest promoting the use of the antidepressant Celexa (citalopram) for use in children and adolescents between 1998 and 2002. The plea agreement imposed criminal fines of $39,500,000 for Celexa’s off-label promotion. Second, Forest entered into a civil settlement agreement to resolve various qui tam False Claims Act lawsuits. The settlement resolved, in part, allegations of fraudulent off-label promotion for both Celexa and Lexapro (escitalopram) for children and adolescents between 1998 and 2005. Forest agreed to pay $149,158,057.66 to settle these claims. Third, Forest entered into a corporate integrity agreement to address Forest’s promotional conduct for a period lasting five years. Each agreement was contingent on the others and each agreement required complete honesty from Forest.
We have been litigating various cases against Forest related to the off-label promotion of Celexa and Lexapro for pediatric use for some years now—inspired by the USAO’s original investigation—in a multidistrict litigation proceeding in the District of Massachusetts. Over the past several years, our litigation has revealed that the scope and extent of Forest’s fraud was not honestly disclosed to the USAO (or, to the Food and Drug Administration) and that Forest misrepresented material facts underlying the USAO’s prosecution. Documents and testimony obtained in our litigation have been unsealed, over Forest’s objection, and we have prepared a 2 detailed memorandum outlining Forest’s misconduct and fraud with the hope the USAO will consider reopening its investigation. Obviously, we are not an unbiased source of information, however, we believe the documents and testimony speak for themselves.
For example, a central feature of Forest’s wrongful conduct, which formed the basis of the government’s investigation, involved the promotion and dissemination of a “positive” Celexa double-blind, placebo-controlled clinical trial in children and adolescents, MD-18, and the suppression of a negative Celexa double-blind, placebo-controlled clinical trial in adolescents, Study 94404. However, unsealed documents and testimony show that the “positive” MD-18 study was not actually positive, and that Forest misled the FDA, the USAO, and the public about this fact. Specifically, MD-18 was only able to achieve a positive result by including nine patients in the study that were, as Forest’s medical director put it, “automatically unblinded” due to a dispensing error. In fact, when the mishap occurred, Forest told the FDA that it would exclude these patients from the primary results. However, when Forest learned it needed the unblinded patients to achieve a positive result, i.e., to show that Celexa outperformed a sugar pill, Forest snuck the patients back into the results, and falsely told the FDA the patients were not actually unblinded.
One internal document, in particular, reveals that this was deliberate fraud. Amy Rubin, a Forest Regulatory Affairs Manager, characterized the dispensing error as having “the potential to cause patient bias” in a draft letter to be sent to the FDA to disclose the unblinding problem. Dr. Charles Flicker, the Senior Medical Director overseeing MD-18, did not approve of this language, stating: “Altho ‘potential to cause bias’ is a masterful stroke of euphemism, I would be a little more up front about the fact that the integrity of the blind was unmistakenly [sic] violated.” Ms. Rubin responded: “Thanks for the compliement [sic]. Part of my job is to create ‘masterful’ euphemisms to protect Medical and Marketing.” Thus, not only was the disclosure to the FDA dishonest, it was, according to a Forest Regulatory Affairs manager, her job to mislead the FDA and protect medical and marketing.
This “masterful euphemism” language was ultimately sent to the FDA. When we deposed Dr. Thomas Laughren, the former official at the FDA who reviewed this study and who, within six months after leaving the FDA, was working as a testifying expert for Forest, he testified that he believed at the time he reviewed the study that there had been no unblinding and, when shown our documents and testimony, that Forest did not honesty disclose the situation to him.
The gravity of this misconduct becomes even more acute when one considers that Lexapro
was ultimately approved by the FDA for adolescent depression in 2009 based on the supposedly
“positive” results of MD-18. Dr. Laughren personally approved the new indication for Lexapro
and testified that, if MD-18 was negative, he would not have approved it. It is our understanding
that this approval weighed heavily into the USAO’s decision to settle their case against Forest.
We urge you to review the enclosed memorandum and accompanying evidence and consider
reopening your investigation of Forest related to its promotion of Celexa and Lexapro for
pediatric use. We would be happy to answer any questions you may have or to meet you in
person to walk you through the evidence.
Sincerely,
BAUM HEDLUND ARISTEI & GOLDMAN, P.C.
By: R. Brent Wisner
MEMORANDUM
How Forest Misled the FDA, USAO, and the Public about the Results of Celexa Study MD-18
January 24, 2018
By:
R. Brent Wisner, Esq.
rbwisner@baumhedlundlaw.com
Michael L. Baum, Esq.
mbaum@baumhedlundlaw.com
BAUM, HEDLUND, ARISTEI & GOLDMAN, P.C.
12100 Wilshire Blvd., Ste 950
Los Angeles, CA 90049
(310) 207-3233
TABLE OF CONTENTS
PART I. EXECUTIVE SUMMARY ............................................................................................. 3
PART II: CELEXA PEDIATRIC CLINICAL TRIALS ............................................................. 10
Celexa Study 94404 Was a Negative Clinical Trial ............................................. 10
Table 1 – Celexa Study 94404 Efficacy Results ................................................... 10
Celexa Study MD-18 Was a Negative Clinical Trial, but Forest Misled the FDA
about the Results ................................................................................................... 12
Table 2 – Celexa Study MD-18 Efficacy Results ................................................. 12
A. General Overview of MD-18 Study .......................................................... 13
B. At the Beginning of the Trial, a Packaging Error Caused Nine Patients,
and their Investigators, to Become Unblinded .......................................... 15
Celexa Study Dispensing Diagram ....................................................................... 18
C. Forest Knowingly Misled the FDA about the Nature of the Unblinding by
Using, As Forest Regulatory Affairs Manager Put It, “Masterful
Euphemisms” to “Protect Medical and Marketing”.................................. 19
D. Despite Misrepresenting the Unblinding to the FDA, Forest Promised to
Exclude the Data from the Patients from Its Primary Efficacy Analysis . 21
E. Forest Reneged on its Promise to Exclude the Unblinded Patients from the
Primary Efficacy Results and, Again, Misrepresented the Unblinding in
MD-18’s Final Study Report .................................................................... 22
F. The FDA Never Fully Considered the Unblinding Issue and a Reasonable
Regulator at the FDA Could Review this New Information and Conclude
Study MD-18 Was Negative ..................................................................... 28
G. Forest Also Misled the FDA about the Results of the Secondary Endpoints
................................................................................................................... 32
Table 3 – Comparison of MD-18 Study Report & Dr. Heart Medical Review .... 35
PART III: FOREST USED FALSE RESULTS FROM MD-18 TO PROMOTE PEDIATRIC
USE OF CELEXA AND LEXAPRO ........................................................................................... 36
PART IV: THE LEXAPRO PEDIATRIC TRIALS .................................................................... 42
Lexapro Study MD-15 Was a Negative Clinical Trial ......................................... 42
Table 4 – Lexapro Study MD-15 Efficacy Results ............................................... 42
Lexapro Study MD-32 Was a “Positive” Clinical Trial for Adolescents, but Did
Not Show a Meaningful Difference between Lexapro and Placebo ..................... 42
PART V: FOREST LEVERAGED THE FALSE RESULTS OF MD-18 TO OBTAIN AN
ADOLESCENT INDICATION FOR LEXAPRO........................................................................ 45
PART VI: FOREST USED THE FALSE ASSERTION THAT MD-18 WAS POSITIVE AND
THE FDA’S APPROVAL FOR LEXAPRO TO NEGOTIATE REDUCED PENALTIES IN
USAO CASE ................................................................................................................................ 53
PART I. EXECUTIVE SUMMARY
In 2010, the USAO in the District of Massachusetts entered into a series of agreements with
Forest Laboratories, Inc. and Forest Pharmaceuticals, Inc., (“Forest”) relating, in part, to the off-label
(unapproved) promotion of Celexa (citalopram) and Lexapro (escitalopram) for use in
children and adolescents.1 Forest agreed to plead guilty to engaging in a misdemeanor count of
off-label promotion for Celexa between 1998 and 2002 and pay $39 million in fines.2
Additionally, Forest entered into a civil settlement agreement to resolve, in part, allegations that
Forest fraudulently induced false claims for the pediatric use of Celexa and Lexapro to be
submitted to government healthcare payers between 1998 and 2005.3 Forest agreed to pay $149
million to settle these claims.4 Forest also entered into a Corporate Integrity Agreement (“CIA”)
designed to monitor the promotional practices of Forest for a period of five years.5 The Plea
Agreement, Civil Settlement, and CIA were conditioned on each other6 and required that Forest
be honest with the USAO about its conduct.7
A central feature of Forest’s wrongful conduct involved the promotion and dissemination of
a “positive” Celexa double-blind, placebo-controlled clinical trial in children and adolescents,
MD-18, and the suppression of a negative Celexa double-blind, placebo-controlled clinical trial
in adolescents, Study 94404. Forest’s one-sided presentation of the efficacy data raised concerns
about how companies such as Forest disclose and use data collected during clinical trials,
particularly when used as part of an off-label promotion campaign. Indeed, the factual claim of
one “positive” trial and one “negative” trial played an important role in the USAO’s prosecution
of the original case against Forest.
Recently unsealed documents and testimony, however, show that Celexa Study MD-18 was
not a “positive study” and that Forest misled the FDA, the USAO, and the public about this fact.
In other words, a material fact that formed the basis of the USAO’s and Forest’s negotiations
was, at that time, false, and Forest knew it. Moreover, this misconduct does not stop there.
Shortly before the USAO and Forest finalized their agreements, the FDA approved Lexapro for use in adolescents, based in part on the misrepresented MD-18 (Celexa) study. The fact that
Forest obtained FDA approval for Lexapro for use in adolescents militated against the
government’s prosecution. The 2009 Lexapro approval, however, was based on the false claim
that MD-18 was positive—a false assertion made to the FDA in 2002 (and reasserted to the FDA
in 2008 as part of Forest’s supplemental New Drug Application (“sNDA”) for Lexapro). If MD-
18 had properly been disclosed as negative, the FDA would not have approved Lexapro for use
in adolescents8 and the government’s prosecution of Forest would have included Forest’s
misrepresentations regarding Celexa’s efficacy in two studies—not just the suppression of Study
94404.
The issue centers on how Forest manipulated the MD-18 data to obtain a “positive” result.
All of the secondary endpoints for MD-18 were negative, meaning Celexa did not outperform
placebo in treating depression on all four of the pre-specified secondary endpoints.9 Moreover,
of those patients who completed the study, i.e., the observed cases, there was also no statistical
difference between Celexa and placebo.10 However, Forest represented to the FDA, USAO, and
others that the primary endpoint for MD-18 was positive because, although the difference was
very small, Celexa appeared to outperform placebo to a statistically significant degree. It turns
out, however, that this “positive” result was based on data from nine patients who were
unblinded during the study. When the data from these unblinded patients is removed, however,
the primary result is negative—indeed, the results are negative across the board on every primary
and secondary endpoint.11
How did this happen? At the beginning of the clinical trial, two clinical investigators
informed Forest that some of their patients were receiving pink pills and others were receiving
white pills.12 This prompted an investigation by Forest, which discovered that a packaging error
had caused the medication for the patients randomized to the Celexa group to be pink, Forest-
stamped, dose-stamped, oval-shaped, commercial Celexa tablets.13 Forest immediately notified
the clinical investigators that the pink pills were commercial Celexa and instructed them to
replace the medication with properly blinded white pills.14 However, for the nine patients
already randomized to the trial, Forest directed the clinical investigators to continue the patients
with the wrong-colored pills.15 This meant the first nine patients were unblinded—the
investigators knew the patients taking the pink pills were taking Celexa and the patients taking
white pills were on placebo.
In March 2000, after Forest learned of the dispensing error but before the results of the study
were known, Forest drafted a letter to send to the FDA explaining the situation. In the original
draft, Forest stated that the dispensing error could have “unblinded the study.”16 However,
Forest’s regulatory affairs manager, Amy Rubin, changed the letter to state that the dispensing
error had the “potential to cause patient bias.”17 This prompted Forest’s medical director, Dr.
Charles Flicker, to respond: “Altho ‘potential to cause bias’ is a masterful stroke of
euphemism, I would be a little more up front about the fact that the integrity of the blind was
unmistakenly violated.”18 Un-phased by Dr. Flicker’s concern that Forest was not being “up
front” with the FDA, Ms. Rubin responded: “Thanks for the compliement [sic]. Part of my job
is to create ‘masterful’ euphemisms to protect Medical and Marketing.”19 For Ms. Rubin,
misleading the FDA was not only acceptable, it was part of her job. And, she did her job well.
The letter sent to the FDA in March 2000 used the masterful euphemism language.20
The letter did, however, state that “[f]or reporting purposes, the primary efficacy analysis
will exclude the . . . potentially unblinded patients[.]”21 Thus, before Forest had the results of
MD-18, Forest recognized the data was corrupted and promised that “[a] full complement of 160
patients” would still be “enrolled under standard double-blind conditions.”22
The results for MD-18 were revealed to Forest in August 2001 and Forest learned, for the
first time, that if the nine unblinded patients were excluded from the analysis, as it had promised
the FDA it would do, the results would be negative.23 But, Forest reneged on its promise to the
FDA. When it submitted the final study report to the FDA in April 2002, Forest included the
unblinded patients in the primary efficacy analysis and buried, in an appendix, the results of the
primary efficacy analysis excluding the unblinded patients.24 In the narrative section of the
report, Forest explained that there had been a dispensing error where nine patients received pink-
colored pills, but the patients “were otherwise blinded.”25 This is in stark contrast to Dr.
Flicker’s unequivocal pronouncement that the integrity of the blind was unmistakenly violated.
In its submission to the FDA, Forest did not disclose that the investigators were unblinded or that
the medication dispensed was Forest-branded, dose-stamped, oval-shaped commercial Celexa
tablets. When the FDA reviewed the results of MD-18, it copied and pasted the language from
the final study report and parroted the claim that pink pills were dispensed but were “otherwise
blinded.”26 Forest’s deception worked—the FDA had no idea that the nine patients were actually
unblinded27 and that the study, when properly analyzed, was negative across the board.
Before the MD-18 study report was even written or given to the FDA, Forest started
promoting the “positive” results of MD-18 to physicians. Forest issued a press release
emphasizing the importance of the positive MD-18 study in a field, i.e., SSRI treatment of
pediatric depression, which had consistently failed to produce positive results28; paid Dr. Karen
Wagner (an investigator on the study) to present the false “positive” results of the study at
various academic conferences and, directly, to physicians in CME programs and in-person offlabel
promotion meetings29; and published the false results of MD-18 in a ghostwritten manuscript and then instructed its sales force to use the publication to promote the use of Celexa
and Lexapro in children.30 None of these presentations and publications disclosed the unblinding
issue. It was buried.
The impact of the off-label promotion of the false data was known to Forest, as demonstrated
in the following slide taken from Forest’s internal marketing plan31 discussing its anticipated
launch of Lexapro:
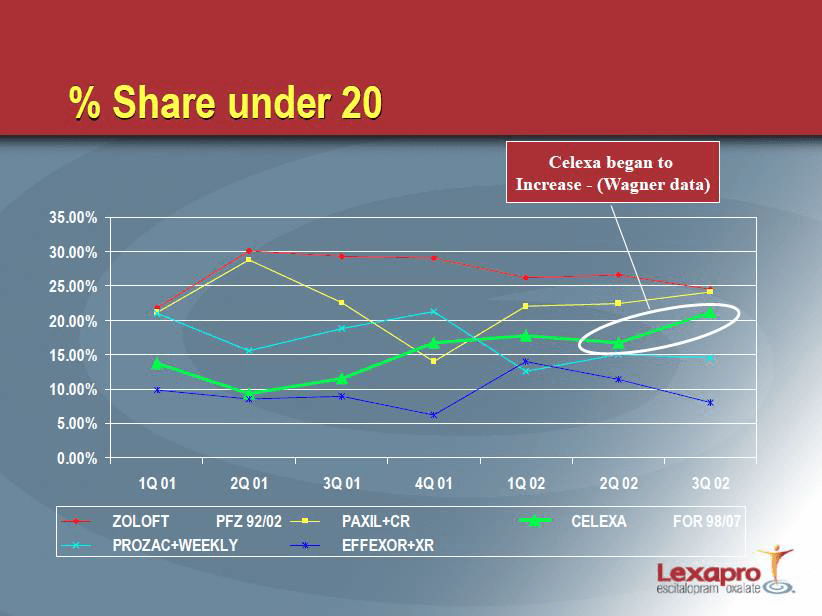
 When Forest’s off-label promotion was finally exposed by the USAO in 2010 and Forest was
When Forest’s off-label promotion was finally exposed by the USAO in 2010 and Forest was
forced to settle and plead guilty to the crime, Forest did not disclose its fraud related to MD-18.
Instead, Forest represented to the USAO and DOJ that MD-18 was positive, militating its
misconduct in suppressing the dissemination of Study 94404. Thus, Forest’s false assertion that
MD-18 was a positive study formed, in part, the basis of the USAO’s negotiated settlements with
Forest. These documents and testimony clearly demonstrate that Forest made material
misrepresentations to the USAO and FDA about this issue and, in fact, continues to do so to this
very day. When Dr. William Heydorn was deposed, the former Forest scientist responsible for
preparing the MD-18 final study report and a named author on MD-18’s publication with Dr.
Wagner, he admitted “I wish we had done things a little differently . . . probably should have
been more forthcoming[.]”32
1 See Exh. 1, DOJ Press Release.
2 Exh. 2, Plea Agreement at 5; see Exh. 3, Criminal Information ¶¶ 55-71 (outlining allegations);
Exh. 5, Side Letter Agreement with Forest Laboratories.
3 Exh. 4, Civil Settlement Agreement at pp.3-4, ¶¶ G(1)-G(3). While the settlement
encompassed the years 1998-2005, there is evidence that Forest’s sales representatives were
illegally off-label promoting Celexa and Lexapro through 2009. One of Forest’s marketing
executives testified: “I have knowledge that representatives may have presented Celexa or
Lexapro inappropriately” and, when asked, “Between 2002 and 2009?” the marketing executive
replied: “Yes.” Exh. 7 Azari Depo at 236:1-237:22.
4 Id. at pp. 6, ¶ 1.
5 Exh. 6, Corporate Integrity Agreement.
6 Exh. 2, Plea Agreement at pp. 11; Exh. 4, Civil Settlement Agreement at pp.3 ¶ E, pp. 10 ¶ 5;
Exh. 6, Corporate Integrity Agreement at 1.
7 Exh. 2, Plea Agreement at pp. 6; Exh. 4, Civil Settlement Agreement at pp.17 ¶ 15.
8 See Exh. 8, 2017 Depo. of T. Laughren at 401:15-402:10 (Dr. Thomas Laughren, the senior
FDA official who approved Lexapro for use in adolescents admitting that he would not have
approved Lexapro for adolescents if MD-18 was negative).
9 Exh. 9, Excerpts of Study MD-18 Rpt. at pgs. 101-104, 244.
10 Id. at 111 (listing p-value of observed cases analysis at week at as 0.1670); Exh. 8, 2017 Depo.
of T. Laughren at 97:1-21, 99:18-21, 343:6-10 (“Q. Sure. But we know that the OC results for
the people who actually completed the clinical trial, that actually was negative for efficacy,
right? A. That’s true.”); Exh. 11, 2016 Depo. of W. Heydorn at 138:24-139:6, 144:6-9.
11 Exh. 12, 2016 Depo. of S. Closter (Forest’s Rule 30(b)(6) Corporate Representative) at
294:10-295:20 (“If they were removed from the study, I understand that the result would have
been negative.”).
12 See Exh. 13, Draft FDA Letter with C. Flicker Handwritten Comments at 1 (describing how
Forest learned of the dispensing error).
13 Id.; see Exh. 14, Memo re. Investigation of CIT-MD-18 Clinical Study Use of Trade-Dress
Citalopram 20 mgs Tabs at 1-2; Exh. 15, Memo re. CIT-MD-18 (Deviation Report) at 1-2 (“It
was brought to our attention . . . some patients enrolled in this study had pink tablets in their bottles. We immediately investigated . . . We discovered . . . the pink oval tablets with FP/20MG
imprints.”).
14 Exh. 16, Email re. CIT-18 FAX to Investigational sites (w/ attachment) at 1 (“[A] copy of the
FAX that went out to all CIT-MD-18 Pediatric Investigational sites this morning is attached[.]”).
15 Id. at 2 (directing patients already randomized to continue on with study).
16 Exh. 17, Email re. Letter to FDA for CIT-18 (w/attachment) at 2 (“The purpose of this letter is
to inform the agency that due to a clinical supplies packaging error for the above-referenced trial,
eight randomized patients at two investigational sites were dispensed medication that could have
potentially unblinded the study.”).
17 Exh. 18, Email responses re. Letter to FDA for CIT-18 at 1.
18 Id.
19 Id.
20 Exh. 19, Letter from T. Varner (Forest) to R. Katz (FDA) at 1.
21 Id.
22 Id.
23 See Exh. 20, Email re. CIT-MD-18 at 1 (“We need to generate Tables 4.1A and 4.1B for ITT
population, excluding the 9 patients who were unblinded at the beginning of the study. Can you
please tell Qiong who they are and try to get the results before 9:30, Friday morning?”).
24 See Exh. 21, Email re. Notes from conference call Oct 4 (w/attachment) at 2 (“[S]ome
citalopram table[t]s were not blinded. The 9 patients who received unblinded medication were
included in the main analyses; a secondary ‘Post-hoc analysis of the ITT subpopulation’ was
done. Refer to these analyses briefly in methods and results and reference the reader to the
appendix table.”); Exh. 9, Excerpts of Study MD-18 Rpt. at pgs. 70, 244 (unblinded results).
25 Id. at pg. 44.
26 Exh. 22, Review and Evaluation of Clinical Data by Dr. Earl Hearst, FDA at 11. All but two
words of Dr. Hearst’s medical review of MD-18 were copied and pasted from the final study
report.
27 Exh. 8, 2017 Depo. of T. Laughren at 154:6-23 (“18 Q Okay. So it was your understanding
that the patients, despite receiving different color tablets, were still blinded, correct? . . . THE
WITNESS: Well, that -- that was -- that was my assumption, correct.”).
28 Exh. 23, 2001 Forest Press Release at 2-3 (“This study is significant because few studies
involving any antidepressant have shown efficacy compared to placebo in the treatment of
depression in children and adolescents . . . Citalopram is now one of the few therapies for which
we have data showing safety and efficacy for this population.” (quoting Dr. Karen Wagner)).
29 Exh. 24, Email re. Ped data at 2 (“[W]e would like to wrap some PR and CME around this
data”); Exh. 25, Email re. ACNP pediatrics abstract at 1 (“John wants GCI to start working a
release and any other way they can spin this data.”); Exh. 26, Emails re. ACCAP meeting at 1 (“You should discuss with GCI bringing her [Dr. Wagner] in for media training prior to the start
of the CME program.”); Exh. 27, Emails re. ACCAP Meeting at 3 (“We spoke with Karen
Wagner today about the current state of affairs regarding the pediatric data. . . She . . . reminded
us that if we want to appeal to the PCP and Pediatric audiences, we need to publish in a place
that provided the appropriate readership . . . She also said that the lack of data regarding the use
of Celexa for pediatrics is limiting it to ‘last choice’ among physicians - she just wanted to make
sure we understood the marketing advantages of the data.”).
30 See, e.g., Exh. 28, Selection of Call Notes at 7, 16-17 (“discussed cx used in children . . . and
results of dr wagner study regarding cx use for children and adolescents . . . Brought up the
Wagner study and sent study to Dr. asked Dr if it would make a difference to use Lx in that age
group since Cx has done well.”).
31 Exh. 29, Lexapro Tactical Presentation at pg. 12; see also id. at pgs. 10-14 (discussing
strategies to increase under 20 market).
32 Exh. 11, 2016 Depo. of W. Heydorn at 307:24-308:15.
PART I: THE PLACEBO EFFECT AND STUDYING ANTIDEPRESSANT EFFICACY
All drugs are susceptible to the placebo effect—the effect a drug has on a patient that has
nothing to do with the medicinal properties of the drug but is caused by the very act of getting
medical attention.33 The belief that one is possibly experiencing medical treatment, by itself, can
create significant and measurable improvement for many conditions.34
In 1962, reeling from news of birth defects caused by a drug called thalidomide, Congress
amended the Food Drug and Cosmetic Act (the Kefauver Harris Amendment, Pub. L. No. 87-
781, 76 Stat. 780 (1962)). Before a drug could be sold as an effective medication, the drug
maker would be required to prove the drug could outperform placebo or, in other words,
demonstrate that the benefit patients receive from a drug could not simply be duplicated by
administration of placebo.35
Today, a drug’s efficacy is determined using double-blind randomized controlled trials
(“DBRCTs”).36 A DBRCT involves the systematic comparison of patients taking a drug and
patients taking a placebo.37 Patients enrolled in the clinical trials are randomly assigned into two
groups. One group takes the drug and the other takes a placebo. However, neither the
investigators nor the patient know which group each patient is in. Once the study is complete,
the benefit observed in the two groups is compared, and if the patients taking the drug
meaningfully outperform the patients in the placebo group, the clinical trial is considered
positive.38 If the drug does not outperform placebo, it is called negative.
As its name suggests, a DBRCT involves three elements, all of which are designed to limit
bias: (1) double-blind (2) randomized (3) controlled trials.39 First, the trial must be double-blind.
This means neither the investigator nor the patient know whether the pill ingested by the patient
is the active drug or placebo.40 If either the investigator or the patient is unblinded, it invalidates
the data since there is no way to determine whether the effects observed are caused by the drug
or caused by the placebo effect (for the patient and investigator). Second, the trial must be
randomized.41 Patients assigned to the drug or control group must be randomly assigned.
Otherwise, the distribution of patients would, itself, inject bias into the study. Finally, the trial
must be controlled. This means the drug must be compared to a control group, i.e., a placebo
pill.42
Before a DBRCT is conducted, a study protocol is generated.43 The protocol specifies the
study’s endpoints—the primary and secondary measures that determine whether the drug
works—and the conduct / procedures of the study. In nearly all DBRCTs, before a study will be
considered positive, the primary endpoint must statistically outperform placebo. This means that
the difference between the drug and placebo must be large enough to conclude the difference
was not a result of chance. Conventionally, and for the purposes of the DBRCTs discussed in
this memorandum, to be considered statistically significant, the endpoint must have a p-value (a
statistical measure) less than 0.05.44
33 Exh. 30, U.S. Food and Drug Administration (FDA), Guidance for Industry, E 10 Choice of
Control Group and Related Issues in Clinical Trials, at 4 (May 2001).
34 Id.
35 See 21 C.F.R. § 314.126.
36 In re Neurontin Mktg. & Sales Practices Litig., 712 F.3d 21, 47-49 (1st Cir. 2013).
37 See FDA, supra note 33, at 4-5.
38 Id. at 5.
39 Exh. 31, Food and Drug Administration (FDA), Guidance for Industry, E9 Statistical
Principles for Clinical Trials, at 10-14 (Sept. 1998).
40 Id. at 10 (“Blinding or masking is intended to limit the occurrence of conscious and
unconscious bias in the conduct and interpretation of a clinical trial arising from the influence
that the knowledge of treatment may have on the recruitment and allocation of subjects, their
subsequent care, the attitudes of subjects to the treatments, the assessment of end-points, the
handling of withdrawals, the exclusion of data from analysis, and so on.”); see FDA, supra note
33, at 4 (listing possible ways bias enters a trial without blinding). In the context of clinical trials
related to depression, this factor is particularly important where a patient’s depression is assessed
by an investigator based on the patient’s answers to specified questions about how they feel. If
either the investigator or the patient knows they are receiving the drug, that knowledge will
likely influence their assessment.
41 FDA, supra note 39, at 12 (“In combination with blinding, randomization helps to avoid
possible bias in the selection and allocation of subjects arising from the predictability of
treatment assignments.”).
42 Id. at 18.
43 Id. at 3 (“For each clinical trial contributing to a marketing application, all important details of
its design and conduct and the principal features of its proposed statistical analysis should be
clearly specified in a protocol written before the trial begins.”). The protocol must be followed
religiously. See Exh. 32, Ravindra B. Ghooi, et al., Assessment and classification of protocol
deviations, 7 PERSPECTIVES CLIN. RES. 3, 132-36 (July-Sept. 2016) (discussing importance of
following protocols and how deviating from them can lead to misleading study results); Exh. 33,
Stephen L. George & Marc Buyse, Data fraud in clinical trials, 5 CLIN. INVEST, 2 161-173
(2015) (discussing how falsification of data, i.e., misrepresenting important events in a clinical
trial, are the most egregious types of misconduct).
44 Exh. 34, Food and Drug Administration (FDA), DRAFT Guidance for Industry, Multiple
Endpoints in Clinical Trials, at 4-5 (Jan. 2017) (discussing the typical use of a p-value of less
than 0.05).
PART II: CELEXA PEDIATRIC CLINICAL TRIALS
There were two pediatric trials conducted on Celexa: Study 94404 and MD-18. And, as
discussed below, both were negative for efficacy on every primary and secondary endpoint.
I. Celexa Study 94404 Was a Negative Clinical Trial
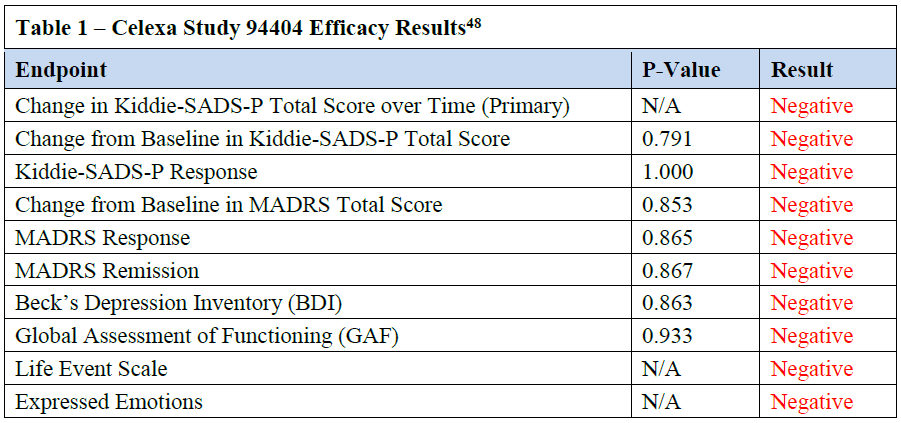
Celexa Study 94404 was conducted in Europe by Forest’s partner, Lundbeck, and was
submitted to the FDA on March 21, 2002.45 The study involved 244 depressed adolescents, aged
13-18.46 The study had one primary endpoint and nine secondary endpoints.47 As illustrated in
Table 1, all endpoints were negative.
 Forest learned that Study 94404 was negative on July 16, 2001.49 This was approximately
Forest learned that Study 94404 was negative on July 16, 2001.49 This was approximately
around the time Forest also learned about the results of MD-18. Forest made a deliberate
decision to suppress the results of 94404 while promoting the results of MD-18.50 Dr. William
Heydorn, a former Senior Medical Writer at Forest, explained:
Q. Were you aware of anyone at Forest Labs who shared the view that it would
be best if the positive data of CIT-MD-18 Was in the marketplace before the
negative data of 94404 was out in the marketplace?
. . .
A. Yes.
. . .
Q. And who did you understand to share that view?
A. I think most of the individuals associated with the citalopram project held that
view.
. . .
Q. Was it your understanding at the time that you were working at Forest Labs
that positive data would be better than negative data in terms of marketing
Celexa?
. . .
A. Yes.
Q. And that positive data being put out in the marketplace over negative data
would be better for the sales of Celexa?
. . .
A. I certainly wasn’t in the sales and marketing department, but that would be my
understanding, yes.51
The investigators at Lundbeck were eager to get the results of 94404 published, but Forest
and Lundbeck wanted to make sure the “positive” results of MD-18 were in the public domain.52
Thus, for three years, the results of Study 94404 remained concealed. Then, in 2004, the New
York Times published an article criticizing Forest for publishing MD-18 without mentioning the
negative results of Study 94404.53 This prompted an immediate response from Forest where it
issued a press release disclosing the results of Study 94404.54 Study 94404, however, did not get
published until 2006—five years after Forest and Lundbeck obtained the results.55
II. Celexa Study MD-18 Was a Negative Clinical Trial, but Forest Misled the FDA about the Results
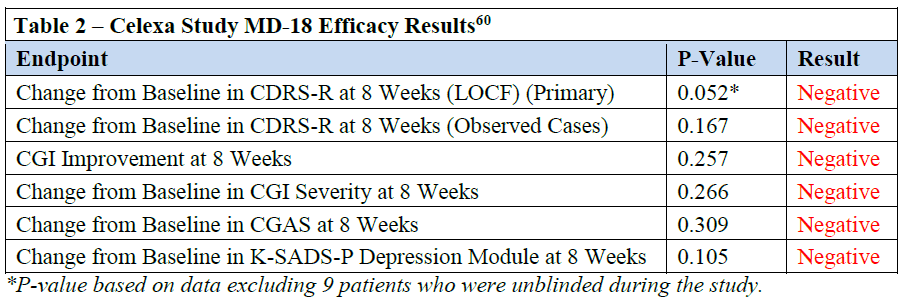
Celexa Study MD-18 was conducted by Forest in the United States. It involved 174 pediatric
patients diagnosed with depression, aged 7-17.56 The final study report for MD-18 was
submitted to the FDA on April 8, 2002.57 MD-18 had one primary and four secondary
endpoints.58 As illustrated in Table 2, all five endpoints were negative. For those patients who
actually completed MD-18, i.e., “observed cases,” the results were also negative (p-value of
0.167).59
In any DBRCT, data collected from the patients must be double-blind. The protocol for MD-
18 stated that: “Any patient for whom the blind has been broken will immediately be discontinued
from the study and no further efficacy evaluations will be performed.”61 Dr. William Heydorn,
the primary author of the final study report for MD-18,62 confirmed that, pursuant to the
protocol, unblinded patients were to be excluded from any efficacy analysis.63 And, this makes
sense. Efficacy in a depression trial is based on an investigator’s subjective assessment of a
patient’s subjective responses to questions on a depression questionnaire. If either the patient or
investigator knows whether the patient is taking a real drug or a placebo, it could very well
influence the patient’s answers and the investigator’s subjective scoring. Indeed, the FDA has
identified multiple types of bias associated with blinding:
- Subjects on active drug might report more favorable outcomes because they
expect a benefit or might be more likely to stay in a study if they knew they
were on active drug. - Observers might be less likely to identify and report treatment responses in a
no-treatment group or might be more sensitive to a favorable outcome or
adverse event in patients receiving active drug. - Knowledge of treatment assignment could affect vigor of attempts to obtain
on-study or follow-up data. - Knowledge of treatment assignment could affect decisions about whether a
subject should remain on treatment or receive concomitant medications or
other ancillary therapy. - Knowledge of treatment assignment could affect decisions as to whether a
given subject’s results should be included in an analysis. - Knowledge of treatment assignment could affect choice of statistical analysis.
Blinding is intended to ensure that subjective assessments and decisions are not
affected by knowledge of treatment assignment.64
Forest told the FDA, DOJ, and USAO that MD-18 was a positive study because, even though
all of the secondary endpoints were negative (as well as the observed cases analysis), Forest
claimed the primary endpoint reached statistical significance. This, however, was untrue. As
discussed in detail below, the first nine patients randomized into MD-18 were unblinded because
of a packaging error. Under the protocol, these initial nine patients should not have been
included in the efficacy analysis. And, without these patients included, MD-18 was negative on
every endpoint, including the primary endpoint. Internal documents and the testimony of former
employees demonstrate that Forest deliberately misled the FDA about the extent of the
unblinding and that MD-18, when properly assessed, is negative.
A. General Overview of MD-18 Study
Dr. Paul Tiseo, Joan Barton, and Dr. Charles Flicker oversaw MD-18.65 Dr. Tiseo was the
Medical Monitor for MD-18, Ms. Barton was the Clinical Trial Manager of MD-18, and Dr.
Flicker was Dr. Tiseo and Ms. Barton’s supervisor, overseeing all of the clinical trial programs
related to Celexa and Lexapro.66 Dr. Tiseo was responsible for the overall conduct of the
study.67 Above Dr. Flicker was Dr. Ivan Gergel and, above him, was Dr. Lawrence Olanoff.68
After MD-18 was completed, Dr. Tiseo left Forest, and Dr. William Heydorn took responsibility
for drafting and publishing the results of MD-18 in both the final study report and subsequent
academic publication.69
When a child was enrolled in the study, the child and their parent were dispensed medication
at different pre-specified intervals as reflected below:
Visit 1 Week -1 Patient dispensed 1 bottle containing 10 placebo pills.
Visit 2 Week 0 Patient randomized. Dispensed 1 bottle containing 10 pills.
Visit 3 Week 1 Patient dispensed 1 bottle containing 10 pills.
Visit 4 Week 2 Patient dispensed 2 bottles containing 10 pills.
Visit 5 Week 4 Patient dispensed 1 bottle containing 40 pills.
Visit 6 Week 6 Patient dispensed 1 bottle containing 40 pills.
Visit 7 Week 8 Study completed.70
At Visit 1 (week -1), each patient was put through a one-week placebo screening period, also
known as a placebo run-in.71 During this period, the patient was given one week of medication
in a 10-pill bottle containing placebo pills.72 This period was single-blinded—meaning the
patient did not know the pills were placebo, only the investigator knew.73
At Visit 2 (week 0), patients were assessed to see how they responded to the 1 week placeboscreening
period.74 If they responded, they were not allowed to enter the randomized portion of
the trial.75 The remaining patients were randomized into either the placebo or Celexa group. At
this point, each patient’s baseline was established.76 The randomization was supposed to be
double-blind, meaning neither the patient nor the investigator knew which group the patient was
assigned to. Each patient was given another 10-pill bottle containing either blinded-placebo or
blinded-Celexa.77
At Visit 3 (week 1), the investigator conducted another round of assessments to see how, if at
all, the patient was responding to treatment.78 As part of this process, each patient was required
to return unused medication and the investigator was required to count the number of remaining
pills in the bottle to ensure compliance.79 At the end of the visit, the patients were dispensed a
new 10-pill bottle to last until the next visit, the following week.80
At Visit 4 (week 2), like at Visit 3, more assessments were done and the pills were counted.
The patients were then dispensed two 10-pill bottles to last the next two weeks.81
At Visit 5 (week 4), the patients were given another round of assessments and the pills were
counted.82 At this half-way point in the trial, the investigators were permitted to increase the
patient’s dose by double if the patient was not responding.83 If so, the patient was expected to
take two pills instead of one each day.84 Accordingly, at Visit 5 (week 4), each patient was given
a 40-pill bottle, which would last two weeks until the next visit.85
At Visit 6 (week 6), more assessments and pill counts were conducted and the patients were
dispensed another 40-pill bottle to last two more weeks.86
At Visit 7 (week 8) the study was completed and the final assessments were performed. The
success of each patient was determined by a comparison of the patient’s improvement (or lack of
improvement) between Visit 2 (week 0) and Visit 7 (week 8).87
B. At the Beginning of the Trial, a Packaging Error Caused Nine Patients, and their Investigators, to Become Unblinded
Shortly after MD-18 began enrolling patients, Forest learned of a packaging error.
According to Dr. Tiseo, two “investigational sites called in to report that some of their patients
were receiving white tablets and others were receiving pink tablets.”88 Forest investigated and,
“it was discovered that a number of bottles of ‘active’ medication were mistakenly packed with
the pink-colored commercial Celexa® tablets instead of the standard white citalopram tablets
used for blinded clinical studies.”89
According to an investigation by the clinical supply group within Forest, the 10-pill bottles to
be used in the Celexa group did not contain the standard white blinded pills, but contained pink,
oval-shaped, Forest-branded, and dose-stamped commercial Celexa® tablets.90 See photo below.

To correct the packaging error, Dr. Tiseo ensured “all sites were notified of this error by
telephone and by fax.”91 In the fax, Dr. Tiseo informed each investigational site about the
packaging error and told each site that the pink pills they were seeing in the patients already
randomized were “pink-colored commercial Celexa® tablets instead of the standard white
citalopram tablets used for blinded clinical studies.”92 Dr. Tiseo explained that “dispensing these
tablets would automatically unblind the study.”93 Dr. Tiseo instructed each investigational site
to immediately return the unblinded medication for repackaging.94 However, for those patients
already randomized, i.e., already receiving the commercial Celexa tablets, Dr. Tiseo instructed
each site to keep using the unblinded medication.95
The problem, however, is that for those patients already randomized into the study, the
patients and the investigators were unblinded. The investigators brought the packaging error to
Forest’s attention because some patients were receiving white pills and some were receiving pink
ones. When Dr. Tiseo told investigators the pink pills were commercial Celexa tablets, even if
the patients somehow did not know the Forest-branded tablets were the active drug, the
investigators knew the patients getting the pink pills were getting Celexa and the patients getting
white pills were getting inert placebos. Dr. Flicker admitted this during his deposition: “[I]f an
investigator were to look . . . at returned medication and he saw that the tablets were pink . . .
then I would think the investigator would be able to draw the conclusion that the patient was on
active drug.”96 Dr. Heydorn also conceded: “If an investigator knows which patients are taking
branded Celexa and which ones are taking white pills, doesn’t that mean the integrity of the blind
was . . . unmistakenly compromised? . . . It does raise questions about the integrity of the blind,
yes.”97
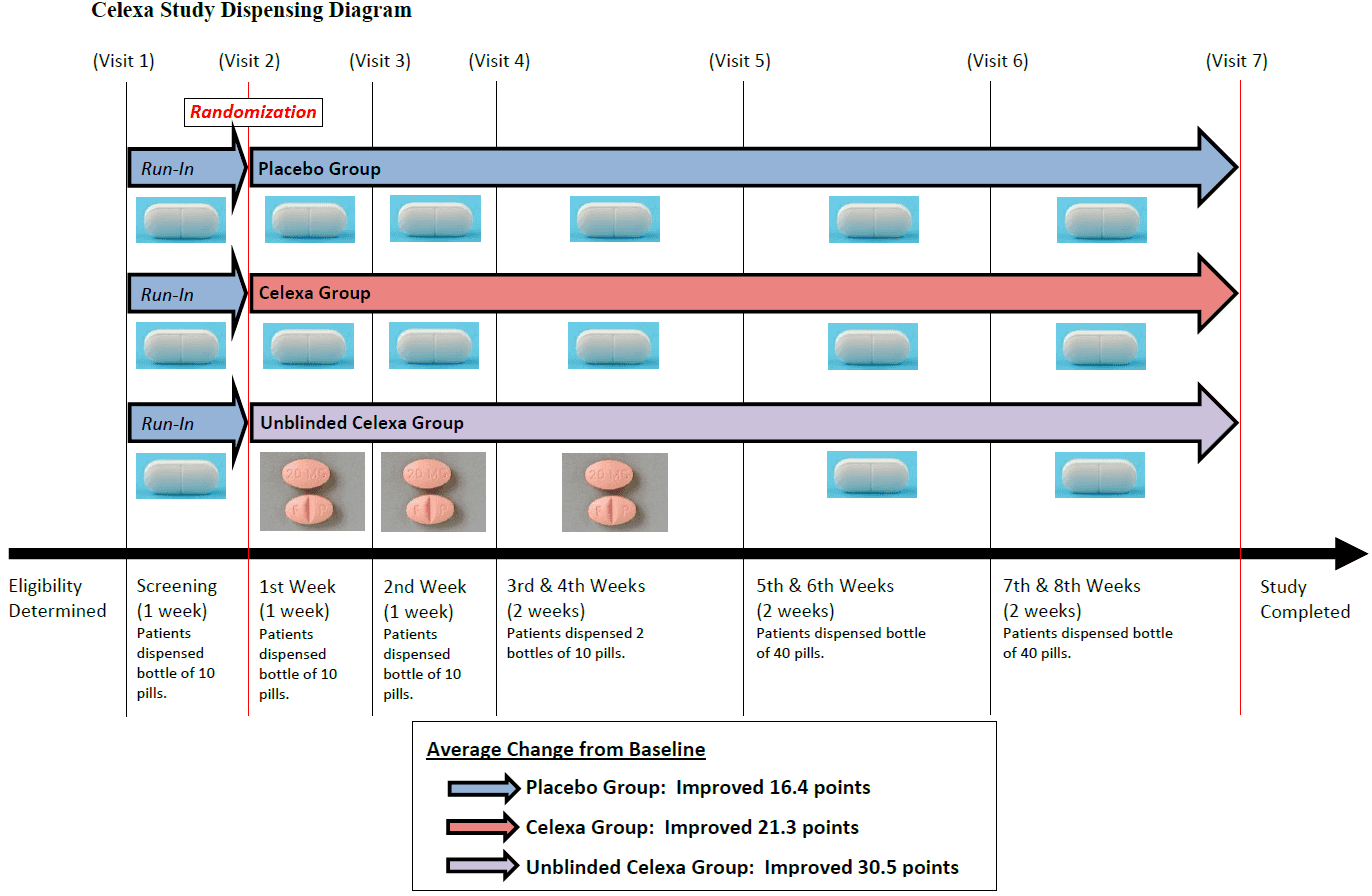
Additionally, there is strong evidence that the patients randomized into the Celexa group
were also, individually, unblinded. First, the average improvement of the blinded patients in the
study taking Celexa was 21.3 points on the CDRS scale.98 However, the average improvement
for the unblinded patients given commercial Celexa for the first four weeks was 30.5 points.99
This 50% greater improvement in the unblinded Celexa patients, versus the blinded Celexa
patients, is strong evidence that those patients or investigators were, in fact, unblinded.
Second, the patients in the Celexa group who were given the commercial Celexa tablets
would have been exposed to different color and shaped pills throughout the trial. Specifically,
the pink tablets were only located in the 10 pill bottles, which were only dispensed during the
four weeks after randomization. The last four weeks of trial used the 40-pill bottles, which
contained the standard, blinded, white pills. However, prior to being randomized, every patient
was given white placebo pills during the one week placebo run-in period. So, this means, these
patients would have been given white pills for one week, pink commercial Celexa pills for four
weeks, then white placebo-looking pills for four weeks.100 This is illustrated in the diagram on
the next page.101

According to the protocol for MD-18, “[a]ny patient for whom the blind has been broken will
immediately be discontinued from the study and no further efficacy evaluations will be
performed.”102 This means any unblinded patients should have been excluded from the study
efficacy evaluations. Dr. Heydorn confirmed this fact: “[P]er the protocol, those patients should
have been excluded because they were unblinded, correct? . . . Yes.”103
Internal Forest documents confirm these patients were, in fact, considered unblinded by the
Forest scientists and statisticians working on MD-18. For example, Ms. Barton sent an email to
Drs. Tiseo and Flicker on December 6, 2000, inquiring about whether MD-18 would need to
have additional patients enrolled due to the fact “the study drug was unblinded.”104 In another
email, dated August 10, 2001, Jane Wu, a biostatistician working on MD-18, explained they
needed to generate tables “excluding the 9 patients who were unblinded at the beginning of the
study.”105 In another email, dated April 5, 2002, Julie Kilbane was finalizing the submission of
MD-18 and sent an email explaining that “[s]ome of the supplies were unblinded for this
study[.]”106 Within Forest, there was no ambiguity about whether these patients were actually
“unblinded.”
C. Forest Knowingly Misled the FDA about the Nature of the Unblinding by Using, As Forest Regulatory Affairs Manager Put It, “Masterful Euphemisms” to “Protect Medical and Marketing”
After correcting the packaging error to prevent further “automatic” unblinding, Forest
debated whether to notify the FDA of the problem. Dr. Tiseo drafted an initial version of a letter
to send to the FDA.107 Dr. Flicker reviewed it and advised not sending any letter, but, if Forest
did send a letter, he advised giving considerably less detail.108 After incorporating Dr. Flicker’s
comments, Dr. Tiseo circulated a draft version of the letter to various Forest executives and
regulatory personnel, including Lawrence Olanoff, Ivan Gergel, Amy Rubin, Tracey Varner,
Julie Kilbane, and Dr. Flicker.109 The draft letter stated that the dispensed medication could have
“unblinded the study.”110 Dr. Tiseo solicited comments from the group.111
Amy Rubin, who worked in Regulatory Affairs, edited the letter, changing the language from
stating that the dispensing error could have “unblinded the study” to stating the error had the
“potential to cause patient bias.”112 Ms. Rubin’s edit drew criticism from Dr. Flicker who felt
Ms. Rubin’s edits were not up front: “Altho ‘potential to cause bias’ is a masterful stroke of
euphemism, I would be a little more up front about the fact that the integrity of the blind was
unmistakenly violated.”113 This criticism, however, did not prompt Ms. Rubin to correct the
language of the letter. Instead, Ms. Rubin responded: “Thanks for the compliement [sic]. Part
of my job is to create ‘masterful’ euphemisms to protect Medical and Marketing.”114 For Ms.
Rubin, misleading the FDA was not only acceptable, it was part of her job.115
And, she did her job well. The letter ultimately sent to the FDA on March 20, 2000,
contained the misleading “masterful euphemism” language.116 The letter did not disclose that the
patients dispensed the pink pills were “automatically unblinded” as Dr. Tiseo stated to the
investigators or that, as Dr. Flicker noted, the integrity of the blind was “unmistakenly violated.”
The smokescreen was up.
As part of the MDL litigation, Plaintiffs deposed Dr. Thomas Laughren, the former Director
of Psychiatric Drug Products in the Division of Neuropharmacological Drug Products at the
FDA. He personally reviewed the final study report for MD-18 while at the FDA and,
ultimately, was the man at the FDA who approved Lexapro for use in adolescents in 2009.117 Dr.
Laughren departed the FDA in 2013 and, within months, was working as a testifying expert for
various pharmaceutical companies, including Forest.118 In fact, Dr. Laughren was hired as a
testifying expert for Forest to provide opinions about the pediatric efficacy of Celexa and
Lexapro and whether the drugs can increase the risk of suicidal behavior in children.119
Notwithstanding Dr. Laughren’s unseemly transition from regulating Forest at the FDA to
working for Forest, he was shown the euphemism emails and other documents, and he took
offense to Forest’s conduct:
[Q]. [D]oes it concern you that the clinical medical director at the time, Dr.
Flicker, believes that a letter that is being proposed to the FDA contains “a
masterful stroke of euphemism”?
[A]. Yeah, no, that’s – that’s concerning, I would say. . . .
[Q.] Does it concern you that an employee for Forest whose job it is to interact
with the FDA states that it’s part of her job to “create masterful
euphemisms to protect medical and marketing”?
[A]. It -- it is objectionable. I mean, my -- my expectation of -- of companies
is that they will be, you know, completely transparent with -- with the
FDA about what happened in the conduct of a trial.
. . .
[Q.] Does it concern you that Ms. Rubin, whose job it was to interact with the
FDA, believes that it’s her job to “create masterful euphemisms to protect
medical and marketing”? . . .
[A.] What -- what concerns me is -- is that -- you know, what was represented
to FDA was not precisely what happened.120
D. Despite Misrepresenting the Unblinding to the FDA, Forest Promised to Exclude the Data from the Patients from Its Primary Efficacy Analysis
While the March 20, 2000 letter to the FDA misrepresented the nature of the unblinding to
the FDA, it also stated: “For reporting purposes, the primary efficacy analysis will exclude the
eight potentially unblinded patients, with a secondary analysis including also to be
conducted.”121 This sentence was added by Dr. Flicker to the original draft of the letter.122 Dr.
Flicker, consistent with his view that the integrity of the blind was unmistakenly violated, knew
the data from these patients was corrupted. So, Forest promised the FDA, consistent with the
express wording of the MD-18 study protocol, that the unblinded patients would not be counted
in the primary efficacy analysis and that, instead, “[a] full complement of 160 patients will be
enrolled under standard double-blind conditions.”123 Dr. Flicker acknowledged: “[Y]ou were
suggesting that the nine patients subject to the dispensing error were not standardly doubleblinded,
correct? . . . I think it does suggest that.”124 Importantly, this promise to exclude the
unblinded patients from the primary efficacy analysis was made before Forest knew the results of
the primary efficacy endpoint turned on the inclusion of those patients.
E. Forest Reneged on its Promise to Exclude the Unblinded Patients from the Primary Efficacy Results and, Again, Misrepresented the Unblinding in MD-18’s Final Study Report
The protocol for MD-18 only required the enrollment of 160 patients.125 However,
accounting for the unblinded patients, the study would need to randomize at least 169 patients
total so that Forest would have a full-complement of patients under standard double-blind
conditions. Forest ultimately randomized 174 patients into the study, including nine of the
unblinded patients.126 So, for purposes of powering the study with sufficient patients, MD-18
did not require the data from the unbinded patients.127
After the MD-18 data was collected, however, Forest reneged on its promise to exclude the
unblinded patients from the primary efficacy analysis. Without any consultation with the FDA,
Forest slipped the unblinded patients into the primary efficacy analysis—combining the data
from the unblinded patients with the blinded cohort—and prepared a secondary “post-hoc”
analysis excluding the patients and put it in an appendix.128 Including these unblinded patients
into the results was “substantial.”129 With the unblinded patients in the study, the primary
endpoint reached statistical significance, but with the unblinded patients excluded—as Forest
promised the FDA—all primary and secondary endpoints were negative. Had Forest done what
it promised, the study would have been negative. In fact, Forest’s corporate representative
conceded that MD-18 is a negative study when the unblinded patients are excluded.130 So did
Dr. Heydorn, the primary author of the MD-18 study report:
Q. By excluding these nine patients, the P-value went from a statistically
significant .038 to a statistically insignificant .052 on the CDRS-R rating
scale after 8 weeks, correct?
[A]. Yes.
Q. So, in other words, this P-value shows citalopram versus placebo was
negative for the primary outcome measure for MD-18, right?
[A]. Yes.131
Indeed, Dr. Heydorn admitted that, if these patients were unblinded, the fact that the study
was negative without them “undermine[s] the assertions that Study 18’s outcome was positive
for showing Celexa significantly improved major depression disorder in children and
adolescents[.]”132 When Dr. Heydorn was shown the internal documents demonstrating that the
integrity of the blind was unmistakenly violated, he conceded Forest was not honest with the
FDA, that Forest misled people about the results of MD-18, and that he would have written the
study report differently:
Q. Do you have any regrets about your involvement with the CIT-MD-18
based on what I’ve shown you today?
A. I wish we had done things a little differently.
Q. Like what?
A. I wish I had known for certain whether the patients, those nine patients
were unblinded, but obviously I don’t know. You showed me a lot of
documents today suggesting that people knew the patients were unblinded.
I don’t know for a fact that they knew that. All I know is what they wrote
on the paper. I wish I was aware of the correspondence with the FDA.
Q. Do you think, based on what I’ve shown you today, that Forest misled
anyone about the results of MD-18?
A. It probably should have been more forthcoming.
. . .
Q. Would you have changed anything in the final study report?
A. If I were the only one involved in writing it, I probably would have
written it somewhat differently.133
When Dr. Heydorn’s testimony was presented to Dr. Laughren, he testified:
Q. It appears based on Dr. Heydorn’s testimony, he did not believe that the
final study report was fully up front or forthcoming with the FDA; isn’t
that true?
[A]. That’s what he’s saying.
Q. And he’s the man who actually was responsible for the final study report
for Study MD-18, right?
[A]. He appears to have been, yes.
Q. Does it concern you that Dr. Heydorn, who was a former FDA employee
himself, thinks that Forest was not as forthcoming as it should have
been with the FDA about its representation of the results from MD-18?
[A]. Yes.134
The original draft of the MD-18 study report was prepared by a company called PharmaNet,
a contract research organization.135 Before the first draft of the report was prepared, Dr. Flicker,
Dr. Heydorn, and two biostatisticians from Forest met with PharmaNet to discuss how the report
should be prepared.136 The notes of the meeting illustrate Forest’s general strategy in dealing
with the unblinded patients:
Dosing error – some citalopram table[t]s were not blinded. The 9 patients who
received unblinded medication were included in the main analyses; a secondary
“Post-hoc analysis of the ITT subpopulation” was done. Refer to these analyses
briefly in methods and results and reference the reader to the appendix table.137
Thus, from the outset, Forest intended to bury the impact of the unblinded data by referring
“to these analyses briefly” and referencing “the reader to the appendix table” on page 244 (of
2,135). It is also worth noting that, even here, in this meeting, Forest was once again stating that
the drugs “were not blinded” and that the 9 patients “received unblinded medication.” As shown
below, this clear admission of unblinding was deliberately removed from the final study report
sent to the FDA.
In the final study report for MD-18, there are four references to the unblinded patients and all
of them are misleading or factually false. The first reference is in a section of the Study Report
titled “Blinding” where it states:
Because of a drug packaging error, the citalopram or placebo tablets initially
dispensed to 9 patients at 3 study centers were distinguishable in color, although
otherwise blinded (see Section 7.0). When this error was identified at the
beginning of the study period, all study medication shipments were replaced in
full with tablets of identical color to remove any potential for unblinding.138
This paragraph is riddled with inaccuracies and misstatements. First, the placebo tablets
initially dispensed were not distinguishable in color—only the Celexa group received the pink
pills, which is why the investigators were unblinded. If both the placebo and Celexa pills had
been pink, then the investigators would not necessarily have known which patients were assigned
to each group. Second, they were not just distinguishable in color—the pink pills were Foreststamped,
dose-stamped, commercial Celexa tablets. The failure of Forest to disclose that the
drug dispensed was commercial branded Celexa is misleading in the extreme. Third, when this
error was identified, the medication for those patients not yet randomized was replaced, but for
the nine patients already in the study, Forest did not replace their medications.139 Forest
instructed each site to continue using the multicolor pills for those patients already randomized to
placebo.140 That means those Celexa patients received white pills for the screening period, pink
pills for the first four weeks, and then white pills for the last four weeks. This paragraph falsely
stated that these patients’ medication was replaced “to remove any potential for unblinding” and
this is simply not true.
Notably, in the original draft of the MD-18 study report prepared by PharmaNet, a section of
the study report contained the language from the original protocol, specifying that “[a]ny patient
for whom the blind had been broken was to be immediately discontinued from the study and no
further efficacy evaluations were to be performed.”141 Dr. Flicker, however, crossed this
language out and inserted the language that ultimately made its way into the final study report:


The second reference to the dispensing error is in the section titled “Changes in the Conduct
of the Study and Planned Analysis” and it reads:
Nine patients (Patients 105, 113, 114, 505, 506, 507, 509, 513, and 514) were
mistakenly dispensed 1 week of medication with potentially unblinding
information (tablets had an incorrect color coating). Therefore, in addition to the
analysis specified in Section 6.4.1 for the primary efficacy parameter, a post-hoc
analysis was performed on an ITT subpopulation that excluded these 9 patients.142
This paragraph is also misleading and factually incorrect. First, these nine patients were not
dispensed one week of medication with potentially unblinding information. These patients
received unblinded drug for the first four weeks, not just one week.143 When this point was
shown to Dr. Heydorn, he admitted the statement about one week was not true: “A. It does say
one week of medication, yes. . . . Q. So that’s not actually true, right, with respect to patients 113
and 513, correct? . . . It would appear not to be true, yes.”144 And, since the investigators were
unblinded, the patients were technically unblinded for the entire study. Second, once again,
Forest stated there was an incorrect color coating, even though the pink pills were actually
Forest-stamped, dose-stamped, commercial Celexa tablets. Third, Forest stated that it was
providing “a post-hoc analysis” excluding these nine patients. But, again, this directly
contradicts the letter Forest sent to the FDA when the unblinding occurred: “For reporting
purposes, the primary efficacy analysis will exclude the eight potentially unblinded patients, with
a secondary analysis including them also to be conducted.”145
Finally, the phrase “potentially unblinding information” is deeply misleading. Ironically, this
was Dr. Flicker’s phraseology—the same person who, back in March 2000, characterized
“potential to cause bias” as a “masterful stroke of euphemism” and felt that the “integrity of the
blind was unmistakenly violated.”146 In the original draft of the MD-18 study report, it stated:
“Nine patients . . . accidently received 1 week of unblinded study drug treatment[.]”147 Note,
there was no “potential” or uncertainty about whether the patients received unblinded study drug
treatment. However, in November 2001, when Dr. Flicker edited the first draft of the report, he
crossed out this language and added the “potentially” language:148

 So, Dr. Flicker stated that the “integrity was unmistkenly violated” in 2000, and then, a year
So, Dr. Flicker stated that the “integrity was unmistkenly violated” in 2000, and then, a year
later, after he learned that the unblinded patients were needed to obtain a positive result on the
primary endpoint, he characterized it as “potentially unblinding information.”
The third reference to the unblinding is in the section discussing the primary efficacy
endpoint. It reads:
Appendix Table 6 presents the results from the LOCF analysis for the change
from baseline to Week 8 excluding data from the 9 patients for whom the study
blind was potentially compromised (see Section 5.3.4). The results from the Week
8 LOCF analysis comparing the mean change from baseline in CDRS-R in the
citalopram and placebo groups was not substantially affected by the exclusion of
those patients; the LSM difference decreased from 4.6 to 4.3 and the p-value
increased from 0.038 to 0.052.149
This is also misleading. And, once again, this is the handiwork of Dr. Flicker. The original draft
of the study report stated: “Appendix Table 6 presents the results from the LOCL analysis for the
change of baseline t week 8 excluding data from the 9 patients . . . who accidently received 1
week of unblinded study drug treatment[.]”150 Dr. Flicker crossed out this language and crafted
some masterful euphemisms of his own:151

 What makes this paragraph so misleading—aside from suggesting these patients were not
What makes this paragraph so misleading—aside from suggesting these patients were not
actually unblinded—is that Dr. Flicker stated that the exclusion of the unblinded patients did not
substantially affect the results of the study. But that is just not true. Excluding the unblinded
patients makes the primary endpoint no longer statistically significant, i.e., negative.152 It
changes the entire result of the endpoint and, by extension, the study. Dr. Heydorn testified:
[Q]. So with the dispensing error patients excluded from the MD-18 primary
efficacy outcome measure, Celexa failed to significantly outperform
placebo in treating pediatric depression, right?
[A]. That appears to be the case.
Q. That would be an important substantial difference, wouldn’t it?
[A]. Yes.153
The final reference to the unblinding was in the section of the report titled, “Validity.”154 It
reads:
The study was designed to provide a valid, prospectively randomized, doubleblind
comparison of the treatment effects of citalopram and placebo. A
medication packaging error partially compromised the study blind for 9 of the 174
patients. Post-hoc analysis excluding these patients supported the results from the
intent-to-treat analysis.155
This section of the report was also drafted by Dr. Flicker.156 And, like the previous sections, it
misstates the effect of excluding the unblinded patients from the trial on the overall results.
Thus, all the sections in the final study report addressing the unblinding issue were drafted by Dr.
Flicker and none of them state, as he previously stated in his email, that the integrity of the blind
was unmistakenly violated. The report was deliberately misleading or, at least in Dr. Flicker’s
own words, not up front.
F. The FDA Never Fully Considered the Unblinding Issue and a Reasonable Regulator at the FDA Could Review this New Information and Conclude Study MD-18 Was Negative
Forest submitted the MD-18 Study Report to the FDA as part of an application seeking a
pediatric indication for Celexa. Ultimately, the FDA denied the application, stating there was
insufficient evidence that Celexa was effective in treating pediatric depression.157 A careful
review of the FDA’s analysis of MD-18, however, reveals that the FDA was misled about the
unblinding situation and, ultimately, the results of the study.
MD-18 was reviewed by Dr. Laughren and Dr. Earl Hearst.158 Dr. Laughren was the Team
Leader of Psychiatric Drug Products and Dr. Hearst was the primary medical reviewer.159
Normally, clinical trials are reviewed by more than one medical reviewer and the FDA conducts
a statistics review, designed to verify the statistics presented by the drug sponsor.160 Dr.
Laughren explained that “[t]he -- the statistical review would likely go into more detail on the --
on the analysis plan and whether or not it was followed in -- in conducting the analysis.”161
However, because the FDA and Forest understood that Celexa would not be approved for
children due to the negative result of Study 94404, the FDA determined “there was no need for a
statistics review of the efficacy data.”162 Instead, the FDA only did a medical review of Study
94404 and MD-18.
Dr. Hearst’s primary medical review of MD-18 concluded, based on the information in the
final study report, that MD-18 was positive.163 Regarding the unblinding issue, Dr. Hearst
copied and pasted the text from the final study report—the report Dr. Heydorn conceded he
would have written differently had he known about the unblinding issue.164 Dr. Hearst copied
verbatim: “[b]ecause of a drug packaging error, the citalopram or placebo tablets initially
dispensed to 9 patients at 3 study centers were distinguishable in color, although otherwise
blinded.”165 Indeed, all but two words of Dr. Hearst’s review of MD-18 consists of sections
copied and pasted from the final study report, suggesting the FDA relied heavily on the accuracy
of the report. And, by copying and pasting from the study report, Dr. Hearst parroted Forest’s
assertion that the data from these unblinded patients was not actually unblinded. Dr. Laughren
acknowledged that Dr. Hearst appeared to have copied and pasted from the final study report and
conceded this was not the approach he endorsed:
Q. And that is a verbatim copy and paste which was in Dr. Hearst’s medical
review, correct?
[A]. Yes . . .That -- that does look like it’s – it’s identical language.
[Q]. Now, Doctor, in the course of your work at the FDA, do you recall
copying and pasting language from a final study report into your medical
review?
A. No, I -- I -- I did not do that.
Q. Why not?
A. Because I preferred to reach my own conclusions.166
Dr. Laughren also prepared a memorandum, which included a review of MD-18.167
Although Dr. Laughren advised against a pediatric indication for Celexa, he stated in reference to
MD-18 that “I agree with Dr. Hearst that this is a positive study in support of the efficacy in
pediatric MDD.”168 With regard to the unblinding issue, he remarked that “[t]here was a
packaging error resulting in tablets being distinguishable for drug and placebo for 9 patients
(although still blinded).”169 When asked about this sentence, Dr. Laughren testified that, based
on the information from the final study report for MD-18, it was his understanding that the
patients received different color pills but were still blinded:
Q. Okay. Now, in that sentence, before that, you said: “There was a
packaging error in tablets being distinguishable for drug and placebo for
nine patients, although still blinded.” It was your understanding that the
patients, despite getting a different color tablet, were still blinded,
correct?
[A]. I – I’m assuming that I made that statement based on something that I had
seen in -- in the supplement.
Q. Okay. So it was your understanding that the patients, despite receiving
different color tablets, were still blinded, correct?
[A]. Well, that -- that was -- that was my assumption, correct.
Q. If in fact the patients were unmistakenly unblinded, that is not what you
understood at the time that you wrote this memorandum, correct?
[A] I -- I -- again, this goes back almost 15 years. I’m not sure what my state
of mind was at the time that I -- that I wrote this memo. But my belief was
based on what I’ve written here is that the patients were blinded.170
Thus, in the absence of clear statements such as “the blind was unmistakenly violated,” Dr.
Laughren believed the study report’s assertion that the patients were not really unblinded and,
thus, their inclusion in the primary endpoint analysis was not a cause for concern. After showing
Dr. Laughren the internal documents where numerous Forest employees stated, in no uncertain
terms, that the nine patients were unblinded, Dr. Laughren agreed that the final study report for
MD-18 misrepresented what happened with regard to the unblinding:
Q. Now, we reviewed the final study report for MD-18. Nowhere in that
study report that we reviewed, the portions that we looked at, did it state
that the integrity of the blind was unmistakenly violated, did it?
A. No.
Q. In fact, the final study report stated that they were otherwise blinded,
didn’t it?
A. It -- it suggests that there was a potential for unblinding, but didn’t
acknowledge that -- that the investigators at least, if they received -- if
they noticed that the tablets had the -- you know, the name “Celexa” on
them and were commercial tablets, that the investigators at least would
have -- would have been unblinded with regard to those patients.171
Dr. Laughren also testified that the information presented to him during his deposition was
“new information” that he did not consider when he reviewed MD-18 at the FDA:
Q. And I’ve also shown you some documents which suggest that Forest
didn’t properly disclose that fact to the FDA in its submissions, correct?
[A]. It -- it certainly would have been my preference that -- that Forest be
more transparent with FDA about the issue of unblinding. . . .
Q. Now, considering that they weren’t transparent about that issue, do you
think – and also in consideration of the fact that Study MD-18 never had a
statistical analysis of the efficacy data, do you think that it would be
appropriate for the FDA to take another look at this data just to make sure
that in fact Study 18 was -- was positive as Forest has represented?
[A]. It -- it isn’t my judgment at this point. . . . So, I mean I -- that – that’s
for FDA to decide at this point. . . . Whether or not FDA -- and I also
told you that, in retrospect, I would have had a statistical review done on
-- on 18.
. . .
And it’s – it’s up to FDA to decide whether or not, you know, based on
this -- on this, you know, new information, which I think is probably
new information from FDA because I wasn’t aware of it at the time.
But it’s not my call.172
According to Dr. Laughren, the information about the unblinding of the patients constituted
“new information” that was not available to him—and was in fact misrepresented to him—while
at the FDA.173 And, even though it was no longer his “call,” Dr. Laughren agreed that a
reasonable regulatory person at the FDA could review this new information and conclude that
MD-18 was negative:
Q. Do you agree, though, Doctor, that a reasonable regulatory person at the
FDA could come to a different conclusion about the positive results of
MD-18?
[A]. It -- this is always a matter of judgment. So the answer would be, yes,
different people looking at the same dataset can reach a different
conclusion.174
G. Forest Also Misled the FDA about the Results of the Secondary Endpoints
Forest did not limit its deception to the unblinding issue—Forest also misled the FDA about
the secondary endpoints in the study. There were four secondary endpoints: (1) GGI-I at 8
weeks; (2) change from baseline in CGI-S at 8 weeks; (3) change from Baseline in CGAS at 8
Weeks; and (4) change from Baseline in K-SADS-P Depression Module at 8 Weeks.175 There is
no dispute that all of the secondary endpoints in the study were negative at 8 weeks, meaning
none of the secondary measures were statistically significant (p<0.05).176 Forest admits they
were negative.177 And yet, in the final study report for MD-18, under the section titled “Efficacy
Conclusions” Forest stated:
Significant differences (p<0.05), indicative of greater improvement in citalopram
patients than placebo patients, were also observed on the CGI-I, CGI-S, and
CGAS. Statistically significant effects were not found as consistently across
study timepoints for the secondary efficacy parameters as for the primary efficacy
parameter, but numerically greater improvement in the citalopram group was
observed on every efficacy parameter at every clinic visit in both the LOCF and
OC analyses.178
This is misleading because it suggests that the CGI-I, CGI-S, and CGAS were statistically
significant “indicative of greater improvement in citalopram” when, in fact, they were all
negative at week 8, i.e., the pre-specified secondary endpoints. Internal documents indicate that
this was a deliberate strategy. Specifically, when Forest contracted with PharmaNet to prepare
the first draft of the MD-18 study report, they had a conference on October 4, 2001, where Dr.
Heydorn, Dr. Flicker, Dr. Jin, and Dr. Wu from Forest attended.179 Notes from the conference
call indicate Forest knew the secondary endpoints were negative, but wanted to spin the data by
focusing on earlier time points in the study when the secondary endpoints were positive:
For secondary efficacy measures – no significant difference at the week 8 LOCF
analysis. The[re] are some significant findings early on in treatment. Forest
looking at individual patient listings to see if there are any clues as to why week 8
findings were not positive. For now, emphasize the positive findings at earlier
time points for the secondary efficacy variables.180
Then, when PharmaNet prepared the first draft of the study report, it stated in the section titled
“Efficacy Conclusions” that:
All other efficacy parameters showed a consistent numerical trend in favor of
citalopram treatment, but failed to reach statistical significance at week 8.
Except for the CGI-I responder score, all other parameters with evaluations at
week 6 reached statistical significance in favor of citalopram treatment at this
timepoint. The by-visit evaluations for these parameters show a marked
improvement in the placebo scores at the week 8-timepoint, suggesting a placebo
effect. No explanation is currently available for this observation. This large
placebo effect may be, in part, responsible for the lack of statistical significance
in favor of citalopram at week 8.181
However, Dr. Flicker crossed out this language and handwrote the language that, for the most
part, ended up in the final study report.182 Notably, Dr. Flicker also attempted to change the
definitions of the secondary efficacy parameters to make them incorporate earlier time points, as
opposed to week 8.183 For example, in the original protocol for MD-18, which Dr. Flicker
signed, it stated that “the endpoints for the secondary objectives are the CGI-Improvement score,
and change from baseline in the CGI-Severity score, K-SADS-P (depression module) score and
CGAS score at Week 8.184” And, PharmaNet faithfully described these objectives in terms of
week 8 in its original draft.185 However, Dr. Flicker crossed out “week 8” in his editing,
suggesting that the endpoint was not at week 8, but at any time period during the study:186




Then, in the section describing the Secondary efficacy parameters, once again Dr. Flicker
crossed out any reference to week 8:187


This elimination of “week 8” from these sections indicates that Dr. Flicker was deliberately
attempting to redefine the efficacy parameters so that Forest’s focus on earlier time points, i.e.,
not at week 8, would appear to be consistent with the study protocol.
The impact of this deception on the FDA’s review of MD-18 is striking. Normally, as Dr.
Laughren explained, the FDA does not pay much heed to the words used in the final study
report: “often when a clinical reviewer gets an application, they often go right to the data rather
than even reading the summary, because they don’t want to be influenced by -- by, you know,
the company’s spin on the data. So they just go right to the datasets and the tables and look at
the data.”188 That, however, did not happen here. Instead, Dr. Hearst, who conducted the
primary medical review of MD-18 lifted, verbatim, the section of the final study report dealing
with the secondary endpoints into his medical review:


This is Dr. Hearst’s only discussion of the results of the secondary endpoints—the only
medical reviewer of MD-18—and it was lifted, verbatim, from Forest’s study report. When Dr.
Laughren was shown this data, he admitted that Forest’s spin on the secondary endpoints had
made its way into the FDA official medical review:
[Q]. Putting Dr. Hearst aside, I’m talking about Forest, we saw that they had a
conference where they said they were going to emphasize this.
A. Yes. Yes. No, it’s -- it is consistent with -- with that view of focusing on
the positive and not giving a complete picture.
Q. And it appears that that spin that Forest put into the final study report
made it into Dr. Hearst’s report, correct?
[A]. It -- it appears to have, yes.189
In Dr. Laughren’s review of MD-18—the only other person within the FDA to review MD-
18—his discussion of the secondary endpoints was even more inaccurate than Dr. Hearst’s. He
stated: “Results also significantly favored citalopram over placebo on most secondary outcomes”
even though every secondary endpoint was negative.190 Dr. Laughren could not remember what
he was thinking when he wrote the statement:
[Q]. Now, on page 3, just above the paragraph that says “comment,” there is a
sentence that reads: “Results also significantly favored citalopram over
placebo on most secondary outcomes.” Do you see that?
A. Yes.
Q. Now, you didn’t state there that all the prespecified secondary endpoints
were negative at week 8, right?
[A]. Correct.
Q. You’re referring here, I assume, to the earlier time points when there were
statistically significant results in the secondary endpoints, correct?
[A]. I -- again, I don’t -- this was written a long time ago. I don’t recall what
would have been in my mind at the time that I wrote this, but it – you’re
correct in saying that it doesn’t -- it doesn’t emphasize the fact that the
eight-week results were all negative on the secondary endpoints.
[Q]. Now, I know you don’t recall this, but is it possible that when you were
drafting this memo, you looked at the final study report, looked at Dr.
Hearst, who you relied upon, and thought, oh, most of the secondary
endpoints must have been positive?
[A]. I -- I would -- I would have to speculate about what -- what I was looking
at at the time when I wrote this, and I -- I -- I prefer not to do that. I just --
I don’t know.
[Q]. Okay. Would you agree with me, though, that it would be accurate to say
all the protocol-specified secondary endpoints for Study MD-18 were
negative at week 8?
[A]. That is -- that appears to be correct, yes.
Q. And would you agree with me that -- that you don’t state that in your
memo?
A. I -- I do not state that in my memo.
Q. And you would agree with me from what we’ve seen in Dr. Hearst’s
clinical review, he did not state that either.
A. He did not appear -- appear to do that either.191
Thus, from the outset, Forest had an objective—avoid disclosing that all the secondary
endpoints were negative and, instead, focus on the secondary measures that reached statistical
significance at various earlier time points. This “spin” was successful. Not only did the primary
medical reviewer at the FDA, Dr. Hearst, copy and paste that spin into his review, but his
supervisor, Dr. Laughren went even further by stating that most of the secondary endpoints
supported efficacy even though they were all negative. Like a cascade, starting with Forest’s
deliberate decision of obscure the secondary endpoints, the deception made its way into the
FDA’s own “independent” reviews.
45 Exh. 35, Excerpts of Study 94404 Rpt. at *1.
46 Id. at *11 (pg. 47 of 345).
47 Id. at *8-10 (pgs. 42-44 of 345).
48 Id. at *12-23 (pgs. 58-69 of 345) (data listed in text).
49 Exh. 36, Letter from Lundbeck to Forest (w/attachment), at 1; Exh. 37, Email re. 94404
Headline results at 1-2 (“94 404 citalopram vs placebo in the treatment of adolescent depression
have been unblinded and unfortunately with a negative result. It was not possible to detect a
significant difference between the two treatment groups.”).
50 See Exh. 3, Criminal Information ¶ 70 (“FOREST PHARMACEUTICALS aggressively
publicized and promoted the results from the positive Forest study, while at the same time
FOREST PHARMACEUTICALS did not publicize or disclose the results of the negative study
to persons outside the FDA or the Danish company which sponsored the negative study. As a
result, doctors and psychiatrists received incomplete and misleading information concerning all
available known data pertaining to the efficacy of using Celexa to treat depression in children
and adolescents.”).
51 Exh. 38, Excerpts of 2007 Depo. of W. Heydorn at 77:23-80:5.
52 Exh. 39, Email re. Publications at 1-2 (“I just wanted to check on the status for the Wagner
pediatric manuscript . . . investigators in the Lundbeck sponsored study seem eager to submit a
manuscript on their study (they are working on it - we have not yet seen any draft) and I wanted
to make sure that the positive data are in the public domain before their negative data get out.”)
53 Exh. 40, Barry Meier, Medicine’s Data Gao – Journals in a Quandary; A Medical Journal
Quandary: How to Report on Drug Trials, NY TIMES (June 21, 2004).
54 Exh. 41, Press Release, Forest Laboratories, Inc., Forest Discusses Disclosure of Citalopram
Clinical Trial Data in Children and Adolescents (June 24, 2004).
55 Exh. 42, Anne-Liis von Knorring, et al, A Randomized, Double-blind, Placebo-controlled
Study of Citalopram in Adolescents with Major Depressive Disorder, 26 J. CLIN.
PSYCHOPHARMACOLOGY 3, 311-15 (2006).
56 Exh. 9, Excerpts of Study MD-18 Rpt. at pg. 38.
57 Id. at pg.1.
58 Id. at pgs. 49-50.
59 Id. at pg. 111.
60 Id. at pgs. 101-104, 111, 244.
61 Exh. 43, Excerpts of Study MD-18 Protocol at pg. 328.
62 Exh. 38, Excerpts of 2007 Depo. of W. Heydorn at 42:13-44:13.
63 Exh. 11, 2016 Depo. of W. Heydorn at 107:13-107:21.
64 Exh. 30, FDA, supra note 33, at 4 (emphasis added).
65 See Exh. 43, Excerpts of Study MD-18 Protocol at pg. 334; Exh. 12, 2016 Depo. of S. Closter
(Forest’s Rule 30(b)(6) Corporate Representative) at 168:1-169:12; 170:5-14; Exh. 11, 2016
Depo. of W. Heydorn at 29:14-32:2.
66 Exh. 43, Excerpts of Study MD-18 Protocol at pg. 334; Exh. 11, 2016 Depo. of W. Heydorn at
29:14-32:2.
67 Exh. 11, 2016 Depo. of W. Heydorn at 29:14-32:2; Exh. 12, 2016 Depo. of S. Closter (Forest’s
Rule 30(b)(6) Corporate Representative) at 168:1-169:12; 170:5-14.
68 Exh. 12, 2016 Depo. of S. Closter (Forest’s Rule 30(b)(6) Corporate Representative) at 168:1-
169:12; 170:5-14.
69 Exh. 38, Excerpts of 2007 Depo. of W. Heydorn at 42:13-44:13; Exh. 11, 2016 Depo. of W.
Heydorn at 29:14-32:2.
70 Exh. 43, Excerpts of Study MD-18 Protocol at pg. 326.
71 Id. at pgs. 323-26.
72 Id.
73 Id.
74 Id.
75 Id. at 318 (“Patients must have a Children’s Depression Rating Scale-Revised (CDRS-R) score
of 40 or greater at both the Screening and Baseline visits.”)
76 Id. at pgs. 323-26.
77 Id.
78 Id.
79 Id.
80 Id.
81 Id.
82 Id.
83 Id.
84 Id.
85 Id.
86 Id.
87 Id.
88 Exh. 13, Draft FDA Letter with C. Flicker Handwritten Comments at 1; accord Exh. 11, 2016
Depo. of W. Heydorn at 197:18-198:14 (verifying that the handwriting belongs to Charles
Flicker); Exh. 44, Emails re. Urgent CIT-MD-18 at *1 (When notified of the findings by a site,
due to seeing white and pink tablets, all supplies were returned and the 10ct bottles re-packaged
with non-trade “white” tablets.” (emphasis added)).
89 Exh. 13, Draft FDA Letter with C. Flicker Handwritten Comments at 1.
90 Exh. 15, Memo re. CIT-MD-18 (Deviation Report) at 1-2.
91 Exh. 13, Draft FDA Letter with C. Flicker Handwritten Comments at 1; accord Exh. 16, Email
re. CIT-18 FAX to Investigational sites (w/ attachment) at 1 (“[A] copy of the FAX that went out
to all CIT-MD-18 Pediatric Investigational sites this morning is attached. All sites have also been
contacted by telephone and given verbal instructions on how to proceed[.]”).
92 Exh. 16, Email re. CIT-18 FAX to Investigational sites (w/ attachment) at 2.
93 Id. (emphasis added).
94 Id. at 4.
95 Id.
96 Exh. 45, 2016 Depo. of C. Flicker at 278:24-279:6.
97 Exh. 11, 2016 Depo. of W. Heydorn at 202:13-19.
98 Exh. 46, J. Jureidini Expert Report at 5.
99 Id.
100 See Exh. 47, J. Glenmullen Expert Rpt. at 24 (discussing how the dispensing process for these
unblinded Celexa patients occurred).
101 Id.
102 Exh. 43, Excerpts of Study MD-18 Protocol at pg. 328.
103 Exh. 11, 2016 Depo. of W. Heydorn at 227:5-10, 228:20-24, 244:11-17.
104 Exh. 48, Email re. CIT-MD-18 Study Drug at 1.
105 Exh. 20, Emails re. CIT-MD-18 at 1.
106 Exh. 44, Emails re. Urgent CIT-MD-18 at *2.
107 Exh. 13, Draft FDA Letter with C. Flicker Handwritten Comments at 1; Exh. 11, 2016 Depo.
of W. Heydorn at 197:18-198:14 (verifying that the handwriting belongs to Charles Flicker).
108 Exh. 13, Draft FDA Letter with C. Flicker Handwritten Comments at 1 (“Reconsider no letter
otherwise I recommend much less narrative[.]”).
109 Exh. 17, Email re. Letter to FDA for CIT-18 (w/attachment) at 1 (“Attached please find the
letter that Charlie and I put together for the purpose of informing the FDA of our packaging
mishap in the citalopram pediatric study.”).
110 Id. at 2 (“[D]ue to a clinical supplies packaging error for the above-referenced trial, eight
randomized patients at two investigational sites were dispensed medication that could have
potentially unblinded the study.”).
111 Id. at 1 (“please review and send your comments back to me within the next few days.”)
112 Exh. 18, Email responses re. Letter to FDA for CIT-18 at 1-2.
113 Id. at 1.
114 Id.
115 Id.
116 Exh. 19, Letter from T. Varner (Forest) to R. Katz (FDA) at 1.
117 Exh. 8, 2017 Depo. of T. Laughren at 401:24-402:8 (“[Y]ou’re actually the one who
ultimately signed off finally on Lexapro's approval for adolescents, right? A Yes.”); Exh. 49,
Lexapro Approval Letter for Adolescents at 3 (signed by Dr. Laughren).
118 Exh. 8, 2017 Depo. of T. Laughren at 81:1-83:14.
119 Id.
120 Id. at 205:14-21, 206:14-23, 207:21-208:4 (emphasis added).
121 Exh. 19, Letter from T. Varner (Forest) to R. Katz (FDA) at 1 (emphasis added).
122 Exh. 13, Draft FDA Letter with C. Flicker Handwritten Comments at 1.
123 Exh. 19, Letter from T. Varner (Forest) to R. Katz (FDA) at 1.
124 Exh. 45, 2016 Depo. of C. Flicker at 294:13-23.
125 Exh. 43, Excerpts of Study MD-18 Protocol at pg. 318 (“The study population will be equally
stratified between children (aged 7 to 11) and adolescents (ages 12 to 17). A total of 160 patients
will be randomized to double-blind treatment.”)
126 Exh. 9, Excerpts of Study MD-18 Rpt. at pg. 64 (“A total of 174 patients received doubleblind
study drug, of whom 89 received citalopram and 85 received placebo.”
127 Id. at pg. 62 (“Assuming an effect size (treatment group difference relative to pooled standard
deviation) of 0.5, a sample size of 80 patients in each treatment group was used[.]”).
128 Id. at pg. 63.
129 Exh. 11, Depo. of W. Heydorn at 88:3-17 (Dr. Heydorn admitting that including the patients
made an “important substantial difference” and that those patients were not needed to power the
study).
130 Exh. 12, 2016 Depo. of S. Closter (Forest’s Rule 30(b)(6) Corporate Representative) at
294:10-295:20 (“If they were removed from the study, I understand that the result would have
been negative.”).
131 Exh. 11, 2016 Depo. of W. Heydorn at 86:22-87:9.
132 Id. at 112:14-112:20.
133 Id. at 308:16-309:6 (emphasis added).
134 Exh. 8, 2017 Depo. of T. Laughren at 263:9-264:5 (emphasis added).
135 Exh. 11, 2016 Depo. of W. Heydorn at 237:2-15.
136 Id. at 236:15-237:6; Exh. 21, Email re. Notes from conference call Oct 4 (w/attachment) at 1
(“Attached are my notes from the conference call with the CRO on the peds study.”).
137 Exh. 21, Email re. Notes from conference call Oct 4 (w/attachment) at 2 (emphasis added).
138 Exh. 9, Excerpts of Study MD-18 Rpt. at pg. 44.
139 Exh. 16, Email re. CIT-18 FAX to Investigational sites (w/ attachment) at 3 (“Patients already
randomized . . . will proceed through the study normally a . . . DO NOT ship their remaining
drug back to Forest. Keep all of their drug at your site and continue to use it as you would
ordinarily. Return only the units of drug for your non-randomized patients.”).
140 Id.
141 Exh. 50, Draft of MD-18 Study Report w/ C. Flicker Comments at 26.
142 Exh. 9, Excerpts of Study MD-18 Rpt. at pg. 63.
143 Indeed, investigators were not notified of the problem until Dr. Tiseo sent out the facsimile on
March 2, 2000. See Exh. 16, Email re. CIT-18 FAX to Investigational sites (w/ attachment) at 1.
At that point in time, three patients had already been in the study for over a month, and the rest
had been in the study for over two weeks. Exh. 9, Excerpts of Study MD-18 Rpt. at pg.1214-15,
1235-37 (listing the dates of each unblinded patients’ various assessments). The statement that
these patients only received incorrectly colored drug for one week is plainly false.
144 Exh. 11, 2016 Depo. of W. Heydorn at 176:3-20 (emphasis added).
145 Exh. 19, Letter from T. Varner (Forest) to R. Katz (FDA) at 1.
146 Exh. 18, Email responses re. Letter to FDA for CIT-18 at 1 (emphasis added).
147 Exh. 50, Draft of MD-18 Study Report w/ C. Flicker Comments at 44 (emphasis added).
148 Id.
149 Exh. 9, Excerpts of Study MD-18 Rpt. at pg. 70.
150 Exh. 50, Draft of MD-18 Study Report w/ C. Flicker Comments at 49 (emphasis added).
151 Id.
152 Exh. 12, 2016 Depo. of S. Closter (Forest’s Rule 30(b)(6) Corporate Representative) at
294:10-295:20.
153 Exh. 11, 2016 Depo. of W. Heydorn at 87:19-88:6.
154 Exh. 9, Excerpts of Study MD-18 Rpt. at pg. 83.
155 Id.
156 Exh. 50, Draft of MD-18 Study Report w/ C. Flicker Comments at 67.
157 Exh. 51, Letter from R. Katz (FDA) to T. Varner (Forest) at 1-2 (“[A] single positive study is
not sufficient, in our view, to support this new claim in pediatric MDD . . . the history of
predominantly negative placebo-controlled trials in pediatric MDD argues against the
extrapolation of the MDD claim from adults to pediatric patients on the basis of the adult data
alone, or even on the basis of one positive study in pediatric patients, along with positive adult
data.”).
158 Exh. 52, T. Laughren, Memo. re. Recommendation for Non-Approval at 1.
159 Id. (“The primary review of the clinical efficacy and safety data was done by Earl Hearst,
M.D., from the clinical group.”).
160 Exh. 8, 2017 Depo. of T. Laughren at 95:10-96:3.
161 Id. at 95:19-22.
162 Note also Laughren Depo. at 94:1-95:6.
163 Exh. 22, Review and Evaluation of Clinical Data by Dr. Earl Hearst, FDA at 2.
164 Compare id. at 11 (“Because of a drug packaging error, the citalopram or placebo tablets
initially dispensed to 9 patients at 3 study centers were distinguishable in color, although
otherwise blinded.”) with Exh. 9, Excerpts of Study MD-18 Rpt. at pg. 44 (“Because of a drug
packaging error, the citalopram or placebo tablets initially dispensed to 9 patients at 3 study
centers were distinguishable in color, although otherwise blinded[.]”).
165 Exh. 22, Review and Evaluation of Clinical Data by Dr. Earl Hearst, FDA at 11.
166 Exh. 8, 2017 Depo. of T. Laughren at 278:9-278:16 (emphasis added).
167 Exh. 52, T. Laughren, Memo. re. Recommendation for Non-Approval at 1.
168 Id. at 3.
169 Id.
170 Exh. 8, 2017 Depo. T. Laughren at 154:6-155:9.
171 Id. at 205:4-13.
172 Id. at 397:9-398:12.
173 Dr. Laughren also testified that he did not believe the new information would have changed
his judgment that MD-18 was a “positive” study, because even though all the secondary
endpoints were negative and the p-value for the primary efficacy endpoint went above 0.05 with
the unblinded patients excluded, he testified it was “close enough.” Id. at 147:7-148:11, 168:21-
169:5. This opinion, however, strains credibility. Back in 2013, before the unblinding issue was
unearthed, Dr. Laughren was deposed as an expert for Forest and he specifically testified that
exclusion of the unblinded patients rendered MD-18 negative: “Q. [I]f these patients were
removed, this would no longer be a positive study? A. That’s correct.” Exh. 53, Excepts of
2013 Depo. T. Laughren at 301:20-302:2 (emphasis added). Indeed, Forest and Dr. Heydorn
both agree that MD-18, with the unblinded patients excluded, is negative. Exh. 12, 2016 Depo.
of S. Closter (Forest’s Rule 30(b)(6) Corporate Representative) at 294:10-295:20 (“If they were
removed from the study, I understand that the result would have been negative.” (emphasis
added)); Exh. 11, 2016 Depo. of W. Heydorn at 87:11-87:14 (same). Dr. Laughren’s “close
enough” opinion is an after-the-fact attempt to justify his conclusion that MD-18 was positive—a
conclusion that formed the basis of his approval of Lexapro for use in adolescents in 2009. To
admit that the study would be negative while excluding the unblinded patients would force him
to concede that he made a mistake in approving Lexapro for use in adolescents.
174 Exh. 8, 2017 Depo. of T. Laughren at 402:18-403:2 (emphasis added).
175 Exh. 43, Excerpts of Study MD-18 Protocol at pg. 321, 329.
176 Exh. 9, Excerpts of Study MD-18 Rpt. at pgs. 101-104.
177 Exh. 12, 2016 Depo. of S. Closter (Forest’s Rule 30(b)(6) Corporate Representative) at 188:4-
20 (confirming, on behalf of Forest that all secondary endpoints were negative).
178 Exh. 9, Excerpts of Study MD-18 Rpt. at 72.
179 Exh. 21, Email re. Notes from conference call Oct 4 (w/attachment) at 1.
180 Id. at 2.
181 Exh. 50, Draft of MD-18 Study Report w/ C. Flicker Comments at 51-52.
182 Id.
183 Id. at 35-36, 38.
184 Exh. 43, Excerpts of Study MD-18 Protocol at pg. 321, 329.
185 Exh. 50, Draft of MD-18 Study Report w/ C. Flicker Comments at 35.
186 Id.
187 Id. at 38.
188 Exh. 8, 2017 Depo. of T. Laughren at 68:18-69:4 (emphasis added).
189 Exh. 8, 2017 Depo. of T. Laughren at 279:6-17 (emphasis added).
190 Exh. 52, T. Laughren, Memo. re. Recommendation for Non-Approval at 3.
191 Exh. 8, 2017 Depo. of T. Laughren at 279:21-282:2 (emphasis added).
PART III: FOREST USED FALSE RESULTS FROM MD-18 TO PROMOTE PEDIATRIC USE OF CELEXA AND LEXAPRO
That Forest off-label promoted Celexa for use in children prior to September 2002 is an
undisputed fact.192 However, starting in December 2001, Forest began aggressively using the
“positive” results of MD-18 to promote the use of Celexa and, later, Lexapro for use in
children.193 Specifically, Forest wanted the data presented at a scientific conference so it could
reference the presentation in various types of promotional activities including continuing medical
education (“CME”) programs.194 On September 21, 2001, John MacPhee of Forest outlines what
he wanted to accomplish with the MD-18 pediatric data: get the pediatric data from MD-18
published as soon as possible so that Forest can use the data in CME programs for marketing.195
A few weeks later, on October 15, 2001, Jeffery Lawrence of Forest wrote to Mary Prescott
at the medical communications company, BSMG/Weber Shandwick, asking about the status of
MD-18 manuscript and explaining that “we would like to wrap some PR and CME around this
data[.]”196 In response, Ms. Prescott explained “I don’t know that any decision has been made
about who is going to write the manuscript (not to be confused with who is going to be the
author[s] of the manuscript. . . . But, for reasons I’ll list below, I think it would make sense to
have a first draft prepared in-house . . . or here, if Bill Heydorn’s group is swamped[.]”197 She
went on to explain that “I’ve heard through the grapevine that not all the data look as great as the
primary outcome data. For these reasons (speed and greater control) I think it makes sense to
prepare a draft in-house that can then be provided to Karen Wagner (or whomever) for review
and comments.”198 She advised that “[r]egarding PR, it will be possible to generate some PR
around the presentation of the data . . . and especially if published in a top-tier journal like
JAMA -- Forest can expect substantial media coverage.”199 This prompted Mr. Lawrence to
email Dr. Paul Tiseo, the researcher overseeing MD-18: “Have you heard anything else about the
Pediatric data? When we last talked, you mentioned some of the measures didn’t look that
great[.]”200
Then, on October 31, 2001, Christina Goetjen of Forest reported back about the company’s
attempt to enlist Dr. Wagner.201 Apparently, Dr. Wagner agreed to help and began advising
Forest about the marketing advantages of the data:
We spoke with Karen Wagner today about the current state of affairs regarding
the pediatric data. . . . She . . . reminded us that if we want to appeal to the PCP
and Pediatric audiences, we need to publish in a place that provided the
appropriate readership (something JAMA would've done.) She also said that the
lack of data regarding the use of Celexa for pediatrics is limiting it to “last
choice” among physicians - she just wanted to make sure we understood the
marketing advantages of the data. I assured her we got it.
She is excited about our Pediatric Regional CME series and will be a fundamental
part of speaker selection. She knows that she and Jeff will be working closely as I
will be on maternity leave.202
In response, Mr. MacPhee explained “my feeling is that the fact that we are last for ped use is
the very reason we can’t wait to disseminate data[.]”203 Ms. Goetjen assured Mr. MacPhee that
Dr. Wagner “is committed to our aggressive timeline as she understands the urgency to get this
data in front of our audience as soon as possible if we’re going to maximize the impact.”204
Ultimately, they all agreed to have BSMG write the first draft: “Bill thought it would be best if
BSMG wrote the first draft... Karen Wagner also realizes that we want this done quickly[.]”205
Forest enlisted Natasha Mitchner, an admitted ghostwriter.206 Notably, a week after these
decisions were made, Mr. Lawarance specifically asked for an Excel file of all “the Celexa
targets who are pediatricians, and or Pediatric Psychiatrists.”207
In December 2001, Dr. Wagner traveled to the annual convention of the American College of
Neuropsychopharmacology (ACNP) in Hawaii and presented the “positive” results of MD-18.208
Her presentation, which was ghost-authored by Ms. Mitchner, did not mention or discuss the
negative results of 94404, it did not discuss the fact that every secondary efficacy endpoint for
MD-18 was negative, and it did not disclose that MD-18’s primary efficacy measure only
achieved statistical significance by including data from unblinded patients.209 Instead, she
presented the “positive” results of the primary endpoint, stating that MD-18 was evidence that
Celexa was effective in children.210
With the “positive” data presented at a scientific conference, Forest immediately started
using the false data to promote the efficacy of Celexa in children. For example, Forest issued a
press release about Dr. Wagner’s ACNP presentation, describing the “positive” results of MD-
18.211 Forest contracted with a company called GCI “to start working a release and any other
way they can spin this data[.]”212 Ms. Goetjen explained that Forest wanted to issue the press
release so it could “put a promotional spin on it and . . . issue additional PR when it is
published[.]”213
In the release, Dr. Wagner is quoted: “This study is significant because few studies involving
any antidepressant have shown efficacy compared to placebo in the treatment of depression in
children . . . Citalopram is now one of the few therapies for which we have data showing safety
and efficacy for this population.”214 The press release made no mention of Study 94404, MD-
18’s negative secondary endpoints, or the unblinding issue, i.e., it had Forest’s “promotional
spin” on it.
Forest then paid Dr. Wagner to travel around the country and tell physicians, in meetings and
formal CME programs, that Celexa was effective in children based on the results of MD-18.
Forest sponsored a CME program which was hosted and presented by Dr. Wagner, where she
cited and discussed the “positive” data from the ACNP presentation to support the message that
Celexa was safe and effective in children.215 Forest sales representatives were specifically
instructed to invite physicians to the CME program—a program for which she played a major
role in selecting speakers.216 And, like her ACNP presentation, her CME presentation did not
disclose Study 94404, the negative secondary endpoints for MD-18, or the unblinding issue.217
Instead, the presentation ended with a multiple choice question: “Which of the following
medications has been shown to be more effective than placebo in the treatment of depression in
children and adolescents?”218 The only available correct answer: Celexa.219 This marketing was
successful, as illustrated in Forest’s marketing plan, showing pediatric prescriptions rose after the
“Wagner data” was promoted.220
In 2004, the MD-18 data was officially published. However, in the years leading up to its
publication, Forest tightly managed how the data was presented. For example, in an email
discussing the company’s publication approach, Dr. Heydorn proposed using a “brief report”
because “[a]s a Brief Report, we feel we can avoid mentioning the lack of statistically significant
positive effects at week 8 or study termination for secondary endpoints.”221 And then again, in
September 2002, the original draft manuscript mentioned a lack of efficacy in children under 12,
but Dr. Heydorn instructed Ms. Prescott to “remove/revise the statement about the lack of
efficacy in children [sic], since the results on that point have been pulled out.”222
When the manuscript finally was published in 2004, it stated that Dr. Wagner was the
primary author and did not disclose the ghostwriters.223 And then, years later, after the criminal
plea by Forest related to this conduct became public, the journal issued a statement about the
Wagner publication, stating that the authors did not properly disclose the fact that commercial
writers were used.224 In the note, it states that Dr. Wagner claimed she was “not aware that Dr.
Heydorn was working with a commercial writer.”225 This was false. There are multiple instances
of Dr. Wagner communicating directly with the ghostwriters. Indeed, Dr. Heydorn confirmed
during his deposition that Dr. Wagner was aware the manuscript was written by ghostwriters.226
To this very day, Forest and Dr. Wagner still cite to and rely on MD-18 as evidence of
Celexa’s efficacy, even though it only achieved a positive outcome by inappropriately including
data from unblinded patients.227 And, Forest was only able to achieve that result by misdirecting
the FDA and misleading the USAO. Indeed, prescribing guidelines for pediatric psychiatry still
provide misleading and false information on Celexa in the treatment of pediatric depression
based on Forest-sponsored publications.
Importantly, the USAO only criminally prosecuted Forest for off-label promotion for Celexa
between 1998 and 2002, and settled civil claims for Celexa and Lexapro through 2005. It turns
out, however, that this conduct continued until, at least, 2009. Gerard J. Azzari was the National
Director of Sales at Forest between 1997 and 2005 and Senior Vice President of Sales between
2005 and 2010.228 Between 1997 and 2010, he oversaw and directed a substantial portion of
Forest’s sales force, i.e., the Forest sales representatives who promoted Celexa and Lexapro to
physicians.229 During his deposition, Plaintiffs asked Mr. Azzari whether it was true that Celexa
was promoted off-label between 1998 and 2002, and he admitted it was.230 Then, he admitted
that this misconduct continued with Celexa and Lexapro through 2009:
Q. So I am going to ask you again, based on your knowledge and experience
between 2002 and 2009, did Forest sales representatives engage in offlabel
promotion of Lexapro for use in pediatric patients?
A. Could I talk to counsel about this question?
Q. Not while it’s pending. I’m asking you for your answer based on your
knowledge and experience. . . . Let me state it again, because I want you to
understand what I am asking you.
A. OK, yes, yes, yes.
Q. Based on your knowledge and experience and your years at Forest,
between 2002 and 2009, did Forest sales representatives engage in offlabel
promotion of Lexapro for use in pediatric patients?
A. I have knowledge that representatives may have presented Celexa or
Lexapro inappropriately.
Q. Between 2002 and 2009?
A. Yes.
Q. And you know that, you have knowledge of that related to Lexapro,
correct?
A. Yes.
Q. And that’s based on your knowledge that child specialists were on
Lexapro call panels between 2002 and 2009, correct?
A. No. My commentary was that individuals may have inappropriately
presented Celexa or Lexapro to physicians.231
192 Exh. 65, Tr. of Arraignment on Information at 16:10-14, 20:18-25 (“MR. STEGER: . . . The
United States would have further demonstrated that beginning in 1998 and continuing thereafter
through at least September, 2002, Forest promoted Celexa for use in treating children and
adolescents suffering from depression, even though Celexa was not FDA approved for pediatric
use. . . . THE COURT: Then likewise the allegations that were made by both counsel, are these
facts true? MR. WEINSTEIN: They’re consistent with what I believe the facts to be. THE
COURT: Okay. So essentially the corporation is pleading guilty to these charges because it is
guilty and for no other reason? MR. WEINSTEIN: That’s correct.”).
193 See, e.g., Exh. 55, Email re. ACP-ASIM at 1 (“[W]e may try a bridging strategy. That is, we
don’t have esc. [Lexapro] data on pediatrics yet, but what if we talk about Celexa and relate it to
Esc. [Lexapro]”); see also Exh. 10, Email re. Stop the Presses at 1-2 (“I believe several of us are
quite anxious to get our hands on this data! When, Bill Heydorn, WHEN ?!!”).
194 Exh. 24, Email re. Ped data at 2 (“[W]e would like to wrap some PR and CME around this
data” . . . Regarding PR, it will be possible to generate some PR around the presentation of the
data at ACNP . . . once the data are presented at a meeting, you can reference that presentation
in other materials. . . ).
195 Id. at 4.
196 Id. at 3.
197 Id. at 1-2.
198 Id.
199 Id.
200 Id.
201 Exh. 27, Emails re. ACCAP Meeting at 2-3.
202 Id. (emphasis added).
203 Id.
204 Id.
205 Id.
206 Exh. 57, Excerpts of Depo. of N. Mitchner at 47:19-48:5.
207 Exh. 54, Emails re. Pediatric Targets at 1.
208 Exh. 56, Email re. Wagner Hot Topics slides (w/attachment) at 1.
209 Id. at 6.
210 Id. at 12.
211 Exh. 23, 2001 Forest Press Release at 2-3.
212 Exh. 61, Emails re. ACNP pediatrics abstract at 1.
213 Exh. 62, Emails re. ACNP Press Releases at 1.
214 Exh. 23, 2001 Forest Press Release at 2-3.
215 Exh. 58, Excerpts of Dr. Wagner’s CME Program at 2-3.
216 See, e.g., Exh. 28, Selection of Call Notes at 7, 16-17 (“discussed cx used in children . . . and
results of dr wagner study regarding cx use for children and adolescents . . . Brought up the
Wagner study and sent study to Dr. asked Dr[.] if it would make a difference to use Lx in that
age group since Cx has done well.”). Plaintiffs are in possession of numerous specific examples
of Forest sales representatives inviting physicians to this CME program, but those call notes are
still, technically, under seal.
217 Exh. 58, Excerpts of Dr. Wagner’s CME Program at 2-3.
218 Id. at 7-8.
219 Id. at 10.
220 Exh. 29, Lexapro Tactical Presentation at pg. 12.
221 Exh. 59, Emails re. Second Draft of Pediatric Manuscript at 1.
222 Exh. 60, Emails re. Citalopram at 1.
223 Exh. 63, Karen Wagner, et al., A Randomized, Placebo-Controlled Trial of Citalopram for the
Treatment of Major Depression in Children and Adolescents, 161 AM. J. PSYCH. 6, 1079-83
(June 2004).
224 Exh. 64, Robert Freedman & Michael D. Roy, Editor’s Note, 166 AM. J. PSYCH. 8, 942-43
(Aug. 2009).
225 Id. at 943.
226 Exh. 11, 2016 Depo. W. Heydorn at 312:24-313:16.
227 See, e.g., Exh. 79, Aaron Levin, Child Psychiatrists Look at Specialty From Both Macro,
Micro Perspectives, 51 PSYCH. NEWS 12, 1-38, 23 (June 2017) (Dr. Wagner referenced and
quoted, espousing the false assertion that MD-18 was positive).
228 Exh. 66, 2016 Depo. G. Azzari at 20:18-21:15.
229 Id. at 21:17-26:5.
230 Id. at 235:7-235:13.
231 Id. at 236:1-237:22 (emphasis added).
PART IV: THE LEXAPRO PEDIATRIC TRIALS
There were two pediatric trials conducted on Lexapro: MD-15 and MD-32. And, as
discussed below, MD-15, the only Lexapro study to test both children and adolescents, was
negative across the board. MD-32, which only tested adolescents, was considered positive from
a statistical perspective, but is suspect from a clinical efficacy perspective.
I. Lexapro Study MD-15 Was a Negative Clinical Trial
The FDA denied the pediatric application for Celexa on September 23, 2002 because:
[T]he results from one of your two studies, Study 94-404, failed to demonstrate
the efficacy of Celexa in pediatric patients with MDD. While we consider the
second study, Study CIT MD-18, to be positive, a single positive study is not
sufficient, in our view, to support this new claim in pediatric MDD. . . . [T]he
history of predominantly negative placebo-controlled trials in pediatric MDD
argues against the extrapolation of the MDD claim from adults to pediatric
patients on the basis of the adult data alone, or even on the basis of one positive
study in pediatric patients, along with positive adult data.232
Shortly after, in December 2002, Forest commenced a double-blind placebo-controlled
pediatric clinical trial of Lexapro: Study MD-15. MD-15 evaluated 264 children and
adolescents between the ages of 6-17.233 And, like Study 94404 and MD-18, every primary and
secondary endpoint was negative for efficacy:
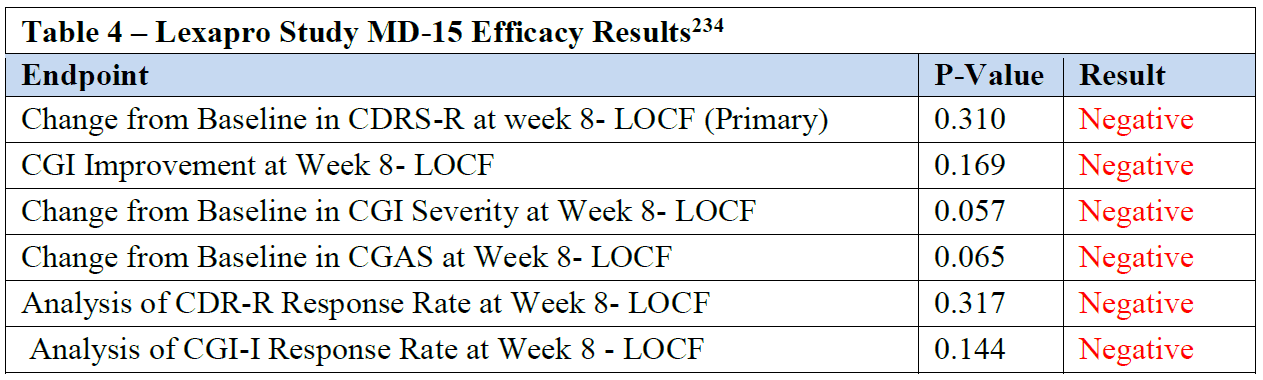
II. Lexapro Study MD-32 Was a “Positive” Clinical Trial for Adolescents, but Did Not Show a Meaningful Difference between Lexapro and Placebo
After Forest obtained the negative results of MD-15 in 2004 (in addition to Study 94404 and
MD-18), Forest was concerned about conducting further pediatric trials. So, Forest obtained an
agreement from the FDA, before the protocol for MD-32 was even written, that if Forest could
obtain a positive result for adolescents in another Lexapro trial, the FDA would approve an
adolescent indication.235 This agreement, however, was specifically contingent on MD-18 being
considered a positive study.236 So, as one Forest executive put it, “everything hinges on SCT-
32.”237
To that end, Forest commenced MD-32 in April 2005, which was completed in May 2007.238
The trial evaluated 316 adolescents (only 259 completed the trial), between the ages of 12-17.239
In stark contrast to every other pediatric clinical trial of Celexa and Lexapro, MD-32 achieved
statistical significance on both primary and secondary endpoints—although the observed cases
analyses on the primary endpoint, i.e., those patients who completed the study, were negative.240
MD-32 has several problems. First, the study was designed to detect a statistical
significance, even with clinically insignificant differences between Lexapro and placebo. It is
widely acknowledged that “[s]tatistically significant effects are not necessarily clinically
meaningful effects.”241 This distinction between statistical and clinical significance exists
because statistical significance is a species of statistics and clinical significance focuses on realworld
effects. Clinical significance is defined as “the smallest difference (i.e., effect size) . . .
that patients perceive as beneficial and that would mandate, in the absence of troublesome side
effects and cost, a change in the patient’s management.”242 Thus, it is entirely possible to
achieve a statistically significant result, even if the difference is trivial, by overpowering a study,
i.e., increasing the sample size. As Dr. Laughren of the FDA acknowledged:
Q. And we discussed earlier that when you increase the sample size in a
clinical trial, what would otherwise be statistically insignificant
differences between the placebo arm and the drug arm can suddenly reach
a statistically significant P-value, correct?
[A]. There’s no question that . . . an increase in the sample size can in some
settings -- it doesn’t always, but it can reduce variance, and therefore, you
know, increase the chance of getting a statistically significant P-value.243
Efficacy in each of the pediatric clinical trials, including MD-32, was measured by
comparing the level of depression, as established using a rating scale, at the beginning of the trial
with the level of depression at the end of the trial. Then, Forest compared the results in the drug
arm with the results in the placebo arm to see if there was any benefit from the drug beyond a
placebo effect. In MD-32, both the Lexapro and placebo patients improved, but the difference in
improvement between the Lexapro and placebo groups was only 3.4 points—out of a scale that
goes up to 113 points.244 This means patients taking Lexapro improved an additional 3.4 points
on the depression rating scale than patients taking a sugar pill. This difference of 3.4 points,
while statistically significant, is so small that no patient or doctor would be able to tell the
difference in real life.245
There are also questions about whether MD-32 was properly conducted. When the patients
were randomized into the study, the Lexapro group started with a baseline score that was
statistically significantly higher than the placebo group, i.e., 1.6 points.246 This indicates there
was selection bias (not true randomization in the Lexapro and placebo groups). On average,
patients in the Lexapro group were 1.6 points worse than the placebo patients, which means there
was more “room” for improvement.247
Forest claims that a difference of 1.6 at baseline is not clinically significant, so it does not
affect the study.248 However, if so, then a difference of 3.4 at the end of the study is also not
clinically significant. One physician who peer reviewed the study manuscript commented: “It
was not clear why the authors consider the baseline difference in the CDRS-R (~2 points)
between the two treatment groups as not clinically significant even though it was statistically
significant. This is confusing as the authors’ then note that a CDRS-R treatment difference
between the groups of ~2pts, which is statistically significant, shows efficacy.”249
Finally, even if one disregards the methodological problems with MD-32, the results hardly
provide substantial evidence of efficacy. There are two primary ways to quantify clinical
significance. The first is called the Cohen effect size.250 “While Cohen defined large, medium,
and small effects as d=0.8, 0.5, and 0.2, respectively, an FDA rule of thumb is that an effect is
deemed large if it is >0.8, small if it is <0.5, and moderate if it falls between those values.”251
The second is known as the number needed to treat (“NNT”). The NNT reflects the number of
people who need to be treated with the drug before one additional person improves more than
taking a placebo.252 “[T]he NNT is a meaningful, well-accepted, common-sense measure[.]” Id.
On the NNT scale, if the number is less than 2, then the drug is considered highly effective. Id.
If the NNT is greater than 4, then it is less effective, since one would need greater numbers of
patients taking the drug before a person fared better than placebo.253
In MD-32, the effect size was 0.27 and the NNT was 8.75.254 These are, by all objective
measures, appalling numbers. This prompted one researcher reviewing Study MD-32 “to
wonder whether the restrictive entry criteria in conjunction with the small effect size limit the
utility of escitalopram in the real world of adolescent MDD. Are these results statistically
significant but clinically not meaningful?”255 And another stated “this is a relatively small ES
[effect size]. Given this small ES, there were no data to see if this level of change had any quality
of life meaning.”256 Considering this is the only statistically positive study for Celexa or
Lexapro, obtained under questionable circumstances, and was limited to adolescents, the results
are small” and unreliable. Standing alone, MD-32 provides scant, if any, evidence of true
efficacy.
232 Exh. 51, Letter from R. Katz (FDA) to T. Varner (Forest) at 1.
233 Exh. 67, Excerpts of MD-15 Study Rpt. at pg. 1 (Initiation date of December 9, 2002).
234 Id. at pgs. 100-105 (showing every primary, secondary, and additional efficacy analyses were
negative).
235 Exh. 68, Letter from FDA re. Forest’s Questions at 1 (“We believe that one additional
positive acute treatment study of adolescents in addition to Study CIT-MD-18 would support a
claim for the acute treatment of adolescents with MDD.”).
236 Id.
237 Exh. 73, Email re. DRAFT Lexapro Road Map at 1.
238 Exh. 69, Excerpts of MD-32 Study Rpt. at 1.
239 Id. pgs. 50, 158.
240 Id. at pg. 158.
241 Exh. 70, Richard S. Keefe, et al., Defining a Clinically Meaningful Effect for the Design and
Interpretation of Randomized Controlled Trials, 10 INNOV. CLIN. NEUROSCI. 5-6 Suppl. A, 4S-
19S (May-June 2013).
242 Id. at 7S.
243 Exh. 8, 2017 Depo. of T. Laughren at 375:5-17.
244 Exh. 69, Excerpts of MD-32 Study Rpt. at pg. 58.
245 See Exh. 47, J. Glenmullen Expert Rpt. at 17 (“But, while statistically significant, the
difference was too small to be clinically meaningful.”).
246 Exh. 69, Excerpts of MD-32 Study Rpt. at pg. 54; Exh. 71, Graham J. Emslie, et al.,
Escitalopram in the Treatment of Adolescent Depression: A Randomized Placebo-Controlled
Multisite Trial, 48 J. AM. ACAD. CHILD ADOLESC. PSYCH. 7, 721-29, 725 (July 2009).
247 Exh. 71, Emslie supra note 246 at 725.
248 Id.
249 Exh. 72, MD-32 Reviewer comments at 8.
250 Exh. 70, Keefe supra note 241, at 11S; accord Exh. 8, 2017 Depo. of T. Laughren at 296:16-
298:12.
251 Exh. 70, Keefe supra note 241, at 11S.
252 Id. at 11S.
253 Id.
254 Exh. 71, Emslie supra note 246 at 726, 727.
255 Exh. 72, MD-32 Reviewer comments at 9.
256 Id. at 8.
PART V: FOREST LEVERAGED THE FALSE RESULTS OF MD-18 TO OBTAIN AN ADOLESCENT INDICATION FOR LEXAPRO
In 2004, Forest sent a request to the FDA inquiring whether “a positive study with
escitalopram using a conventional acute treatment design . . . along with the previous positive
study with citalopram (Study CIT-MD-18) be adequate to support an indication for acute
treatment in pediatric patients aged 12 - 17 years?”257 Relying on the false claim that MD-18
was positive, Forest wanted to know whether an additional positive study in adolescents would
be enough to obtain an adolescent indication for Lexapro. In response, the FDA stated: “We
believe that one additional positive acute treatment study of adolescents in addition to Study
CIT-MD-18 would support a claim for the acute treatment of adolescents with MDD.”258 Thus,
if Forest could obtain a positive adolescent clinical trial for Lexapro, the FDA agreed to give
Lexapro an adolescent indication. This promise, however, was based on a belief that MD-18 was
a positive study. As Dr. Laughren explained:
Q. Okay. All right. So my understanding based on the response from the FDA
is that if Forest could produce a positive double-blind, placebo-controlled
clinical trial with Lexapro in children aged 12 to 17, it would then agree to
provide an indication for Lexapro for that age group.
A. Yes, that’s -- that is what it’s saying. I mean, of course, it would -- you
know, it would have to be reviewed. It’s subject to review by FDA. But
in principle, yes, that is what this letter says.
. . .
Q. Okay. If MD-18 was negative -- okay, just assume that for a second --
would the FDA have made this agreement? . . .
[A]. No. I don’t -- I don’t believe so. That would be my impression that – that
we would not have -- have reached that agreement.259
Ultimately, Forest was able to obtain a statistically positive result in MD-32, as discussed
above. Forest then, in 2008, submitted the results of MD-32 and MD-18 as part of a
supplemental new drug application seeking an adolescent (12-17) indication for Lexapro. Since
the FDA had already promised to approve the application, its review of the data was barebones.
However, before delving into the FDA’s review of this supplemental application, it is
important briefly to discuss the FDA’s original review of MD-18, again. MD-18 involved both
children (7-11) and adolescents (12-17).260 However, the study was not powered to measure just
children or adolescents.261 And, after getting the results, Forest conducted several statistical
analyses of the data and stated, repeatedly, that “[s]imilar effects were seen in the children and
adolescent subgroups, as evidenced by the lack of a treatment-by-age group interaction[,]” that
“the magnitude of the treatment effect was similar in the child and adolescent subgroups[,]” and
that “[t]he magnitude of the mean citalopram-placebo differences on the efficacy ratings was
numerically higher in the adolescents than in the children, but no significant treatment-by-age
group interactions were observed, indicating that the citalopram treatment effect was not age
dependent.”262 And, this lack of treatment-by-age difference was seen across every primary and
secondary endpoint.
Despite this, when Dr. Laughren prepared his memorandum in 2002—when both Forest and
the FDA knew the FDA would not be approving a pediatric indication for Celexa263—he noted
that the observed treatment effect in the study was primarily coming from the adolescent
subgroup.264 When Dr. Laughren was asked about this during his deposition, he explained “[i]t’s
something that I -- that I generally do. I -- you know, I explore a little bit more.”265 It was, as he
put it, an “exploratory” analysis and was not a prespecified hypothesis.266 Thus, there was no
statistical analysis done on the data to determine whether the difference between children and
adolescents was statistically significant:
Q. And so just based on what you said here, do you know whether or not the
differences observed here were statistically significant or not?
A. I -- I don’t. And again, from my standpoint, it -- it wouldn’t be that
important. Because a P-value, whether it met that usual threshold of
statistical significance would not be particularly relevant for something
that wasn’t -- that wasn’t being prespecified and tested.
I mean -- and you could do that. You could say if you make it on the
overall analysis, then you get to -- you have another 0.05 to look first at --
at adolescents, and if you win there, then you get to look at -- but it wasn’t
done that way.267
It is also important to note, as discussed above, that when MD-18 was reviewed by the FDA,
there was no statistical review done on the study268 and Dr. Hearst, quite literally, copied and
pasted his entire MD-18 analysis from Forest’s final study report for MD-18.
However, when MD-18 was resubmitted to the FDA in 2008 as part of Forest’s application
for an adolescent indication for Lexapro—an application the FDA had already agreed to approve
before Forest even started enrolling patients in MD-32—the FDA did not conduct any significant
rereview of the data, but relied exclusively on Dr. Hearst’s and Dr. Laughren’s prior reviews of
MD-18.
Specifically, the primary review of the supplemental application was done by Dr. Roberta
Glass,269 the team leader review was done by Dr. Ni Khin,270 the statistical review was done by
George Kordzakhia,271 and the overall review and final approval was issued by Dr. Laughren.272
Dr. Glass, as the primary reviewer, did not conduct any in-depth analysis of MD-18. Rather,
Dr. Glass assumed that MD-18 was already positive, as she specifically noted that “[t]he sponsor
reached an agreement with FDA that a pediatric claim for escitalopram, an isomeric version of
citalopram, could be obtained with the support of one positive pediatric study in escitalopram in
addition to the one positive study in citalopram.”273 According to Dr. Laughren, Dr. Glass was
focused only on MD-32, not MD-18:
Q. All right. So it appears that Dr. Glass is operating off of the fact that Study
MD-18 was positive and that they just had to look at whether or not there
was an additional positive study for adolescents with Lexapro; is that
right?
. . .
[A]. That’s correct.274
This superficial approach to reviewing MD-18 is reflected in her report. Dr. Glass simply
copied and quoted Dr. Laughren’s analysis, which included data from the unblinded patients,
stating: “The study is positive for the primary efficacy variable of change from baseline of the
CDRS-R total Score (p=0.038). . . . As Dr. Laughren noted in his memo of 9/16/02, ‘…it appears
that the positive results for this trial are coming largely from the adolescent subgroup.’”275
Indeed, Dr. Glass copied and pasted Dr. Laughren’s exploratory analysis into her review.276 And,
like Dr. Laughren, there is no statistical analysis of the difference between children and
adolescents or any discussion about the fact the study report specifically stated that the
differences were not driven by age. Dr. Glass also never mentioned or discussed the effect of the
unblinding—indeed, there is no indication she was even aware of it or its significance. When
Dr. Laughren was shown this data during his deposition, he agreed:
Q. It appears that Dr. Glass is relying on your exploratory analysis of the
different effects observed in the pediatric and adolescent subgroup in your
memo of September 16th, 2002.
A. That’s correct.
Q. And indeed, she has pasted the results on the next page. It says “Summary
of Primary Efficacy Variable for Study 18 by Age Subgroups,” and it says
-- literally says: “Extracted from memorandum by Laughren, September
16, 2002.” Do you see that?
A. I do.
. . .
Q. It does not appear that she did a comprehensive clinical review of MD-
18 at this point; is that right?
. . .
[A]. That’s likely the case, yes.277
And, Dr. Khin likewise conducted a superficial review of MD-18—again, predicated on the
fact that the FDA had already determined that MD-18 was positive. Dr. Khin explained: “In this
review cycle, our review of efficacy was focused on the positive results from one placebo
controlled short-term study (study SCT-MD-32) in our evaluation of the efficacy and safety of
escitalopram in the acute treatment of MDD in adolescents.”278 Dr. Laughren explained:
Q. Would it be fair to say that they had marching orders at this point in their
review that Study MD-18 was positive, just look at 32 and tell us if that’s
also positive?
[A]. I -- I don’t -- I don’t know that I would call that marching orders. . . . I
think there was -- there was that understanding that, you know, we had
already looked at -- at 18 and made a judgment that it was a positive study.
I mean, certainly no one instructed them not to look at 18. . .
Q. . . . [T]hey appeared at least to have been relying upon the agreement that
the FDA reached with Forest in 2004.
A. I think that’s fair.279
Consistent with that approach, Dr. Khin specifically stated that she relied on Dr. Hearst’s (the
FDA reviewer who copied and pasted from Forest’s final study report) and Dr. Laughren’s
reviews: “I would refer to the clinical review by Dr. Earl Hearst dated 9/12/02 and a
memorandum by Dr. Thomas Laughren dated 9/16/02 regarding their reviews of materials
submitted under supplemental NDA for citalopram on 04/18/2002. I will briefly summarize their
interpretation of results from the Study 18 . . . below.”280 Then, Dr. Khin proceeded to copy and
paste Dr. Laughren’s exploratory analysis from 2002—the very analysis that was never
subjected to any statistical analysis.281 And, she made her conclusion relying on the Hearst and
Laughren analyses: “Based on prior clinical review by Dr. Hearst and Dr. Laughren’s memo, we
should be able to count on positive efficacy results from citalopram study 18 in the same aged
population for acute treatment of MDD.”282 Dr. Laughren confirmed this:
Q. So it appears that Dr. K[h]in is relying heavily, if not exclusively, on Dr.
Hearst and yourself’s analysis of Study MD-18.
[A]. That’s correct. Now, of course, this is the team leader review. It's not the
primary review.283
This reliance on Dr. Laughren’s exploratory review was raised during his deposition as well:
Q. When you prepared your memo for CD – for MD-18, and you did this
exploratory analysis dividing the adolescents from the children, did you
anticipate that that being -- that was going to be used to support an
indication for a different drug in adolescents? . . .
[A]. I -- I doubt that I was thinking ahead that far.
Q. Fair enough. In retrospect, it seems that that’s exactly what happened.
A. That’s true.284
Finally, Dr. Kordzakhia conducted the FDA’s statistics review for the supplemental
application.285 But, that statistics review, like Dr. Glass’s and Dr. Khin’s reviews, was limited to
MD-32.286 This means, for a second time, MD-18 evaded any meaningful statistical review. Dr.
Laughren admitted this was not typical:
Q. Is that typical for a pivotal trial that's going to be used to support
indication to have just not been given any statistical review? . . .
[A]. It’s prob- -- it’s probably not typical.
. . .
Q. Do you think that probably would have been helpful, particularly since
you’re using a particular subgroup of an exploratory analyses that you did
in your review of the study? . . .
[A]. In -- in retrospect, I think I -- I would have preferred that.287
Ultimately, Dr. Laughren approved the supplemental application and Lexapro was indicated
for adolescent depression in March 2009.288 And, subsequently, Forest continued to aggressively
market and sell Lexapro for use in adolescents throughout the United States, albeit now it was
“legal.” Dr. Laughren, however, admitted that, if MD-18 was a negative study, he would not
have approved the adolescent indication for Lexapro:
Q. If MD-18 was in fact negative, would you ever have approved Lexapro for
use in adolescents? . . .
[A]. I mean, if -- if -- if you couldn’t rely on 18 as a source of evidence, then
you would’ve only had one source of evidence for Lexapro. So the answer
is this is speculation, but I -- I would not have recommended approving it.
. . .
Q. You’re the one who ultimately did approve it, right?
A. Because I -- I considered Study 18 a reasonable source of evidence.
Q. No, I know. And I’m just saying it’s not speculation because you’re
actually the one who ultimately signed off finally on Lexapro’s approval
for adolescents, right?
A. Yes. . . .
Q. And you’re saying you wouldn’t have approved it if there was only one
study, positive Study 32, right? . . .
[A]. That’s correct.289
And, while Dr. Laughren defended his decision to approve Lexapro for adolescents during his
deposition, he admitted that the FDA, even today, could re-review this data and conclude that
MD-18 was negative and, thus, rescind the Lexapro approval for adolescents:
Q. Do you agree, though, Doctor, that a reasonable regulatory person at the
FDA could come to a different conclusion about the positive results of
MD-18? . . .
[A]. It -- this is always a matter of judgment. So the answer would be, yes,
different people looking at the same dataset can reach a different
conclusion.290
When one considers that Forest’s corporate representative admitted, under oath, that MD-18 is
negative when the unblinded patients are removed,291 and the mountain of evidence showing
these patients were, in fact, unblinded,292 the scope of the fraud comes into view—by misleading
the FDA about the unblinding in MD-18 using “masterful euphemisms”293 and deliberately
misleading language, Forest bamboozled the FDA into approving Lexapro for use in adolescents.
And, now, the man responsible for allowing this mischarge of science—indeed, the godfather of
all modern antidepressants,294 Dr. Laughren—started a company called Psychopharm
Consulting, where he touts having “29 years of experience at the FDA in assisting
pharmaceutical companies with psychiatric drug development programs,” and “hope[s] to
continue in this effort as an independent consultant.”295
257 Exh. 68, Letter from FDA re. Forest’s Questions at 1.
258 Id.
259 Exh. 8, 2017 Depo. of T. Laughren at 368:24-369:9.
260 Exh. 9, Excerpts of Study MD-18 Rpt. at pg. 38.
261 Exh. 21, Exh. 21, Email re. Notes from conference call Oct 4 (w/attachment) at 2 “Note that
study was not powered to look at differences within the two subgroups (children and
adolescents).”
262 Exh. 9, Excerpts of Study MD-18 Rpt. at pgs. 69, 72, 83.
263 Exh. 8, 2017 Depo. of T. Laughren at 94:23-95:4 (“Q. Would it be fair to say then that when
you stated here that the agreement between the sponsor and FDA that these trials were negative
was referring to negative in the sense that it wouldn’t be sufficient to secure a pediatric
indication? A. That’s – that’s the way I interpret that, yes.”).
264 Exh. 52, T. Laughren, Memo. re. Recommendation for Non-Approval at 3 (“[I]t appears that
the positive results for this trial overall are coming largely from the adolescent Subgroup.”).
265 Exh. 8, 2017 Depo. of T. Laughren at 283:5-8.
266 Id. at 284:10-21.
267 Id. at 284:24-285:14.
268 Exh. 52, T. Laughren, Memo. re. Recommendation for Non-Approval at 1 (“Since there was
agreement between the sponsor and FDA that these trials were negative, there was no need for a
statistics review of the efficacy data.”).
269 Exh. 74, Roberta Glass, Clinical Review, FDA.
270 Exh. 75, Ni Khin, Memorandum re. Recommendation of Approval Action for Lexapro, FDA.
271 Exh. 76, George Kordzakhia, Statistical Review and Evaluation, FDA at 1, 3 (indicating that
the statistical review were done on MD-32)
272 Exh. 77, T. Laughren, Recommendation for Approval Action for Lexapro, FDA; Exh. 49,
Lexapro Approval Letter for Adolescents at 3.
273 Exh. 74, Glass supra note 269 at 8; see also id. at 9 (“On November 16, 2004, the Division
confirms that one additional positive acute treatment study with escitalopram in adolescents, in
addition to Study CIT-MD-18, is adequate evidence to support a labeling claim that escitalopram
is an effective acute treatment of MDD in adolescents. Thus, Study SCT-MD-32 in adolescent
patients was initiated in February 2005.”).
274 Exh. 8, 2017 Depo. of T. Laughren at 381:6-12.
275 Exh. 74, Glass supra note 269 at 22.
276 Id. at 23.
277 Exh. 8, 2017 Depo. T. Laughren at 382:13-383:16 (emphasis added).
278 Exh. 74, Khin supra note 270 at 2.
279 Exh. 8, 2017 Depo. of T. Laughren at 387:5-388:2.
280 Exh. 74, Khin supra note 270 at 3.
281 Id. at 6 (“Summary of primary efficacy results by age group for Study CIT-MD-18 - LOCF
(data extracted from Dr. Laughren’s memo dated 9/16/2002)”; see id. at 5 (“Please see Table 2 in
section 5.1.3 regarding summary of primary efficacy results by age group for Study CIT-MD-18
(LOCF data extracted from Dr. Laughren’s memo dated 9/16/2002)”).
282 Id. at 6-7.
283 Exh. 8, 2017 Depo. of T. Laughren at 389:12-18.
284 Id. at 392:12-393:1 (emphasis added).
285 Exh. 76, Khin supra note 270.
286 Id. at 1, 3 (indicating that the statistical review were done on MD-32).
287 Exh. 8, 2017 Depo. of T. Laughren at 384:4-385:7.
288 Exh. 49, Lexapro Approval Letter for Adolescents at 3, at 1-2.
289 Exh. 8, 2017 Depo. of T. Laughren at 401:15-402:16 (emphasis added).
290 Id. at 402:18-403:2 (emphasis added).
291 Exh. 12, 2016 Depo. of S. Closter (Forest’s Rule 30(b)(6) Corporate Representative) at
294:10-295:20 (“If they were removed from the study, I understand that the result would have
been negative.”).
292 Exh. 16, Email re. CIT-18 FAX to Investigational sites (w/ attachment) at 2 (“[D]ispensing
these tablets would automatically unblind the study.”); Exh. 18, Email responses re. Letter to
FDA for CIT-18 at 1 (“[T]he integrity of the blind was unmiatakenly violated.”); Exh. 20, Email
re. CIT-MD-18 at 1 (“We need to generate Tables . . . excluding the 9 patients who were
unblinded at the beginning of the study.”); Exh. 21, Email re. Notes from conference call Oct 4
(w/attachment) at 2 (“[S]ome citalopram tables were not blinded. The 9 patients who received
unblinded medication were included in the main analyses[.]”); Exh. 11, 2016 Depo. W. Heydorn
at 155:2-24, 157:18-21, 218:6-13 (“Q. So with respect to the nine patients who received the pink
tablets, the study was unblinded with respect to them automatically, correct? . . . THE
WITNESS: I guess yes. . . . Q. So they were unblinded as well, correct? . . . THE WITNESS:
With respect to those patients, I would assume so. . . Q. Now, having seen this e-mail from Dr.
Flicker and the fax from Dr. Tiseo, would you agree that the patients who were subject to the
dispensing error were actually unblinded? . . .THE WITNESS: I don’t know for a fact, but that’s
the implication from these letters, yes.”); Exh. 46, J. Jureidini Expert Report at 5 (showing that
the results from the unblinded Celexa patients were 50-60% greater in than blinded Celexa
patients).
293 Exh. 18, Email responses re. Letter to FDA for CIT-18 at 1.
294 Exh. 8, 2017 Depo. T. Laughren at 27:23-28:7 (“Would it be fair to say that during your time
at the FDA, you were involved in some capacity with the approval or review of all of those
SSRIs? A. Every one of them[.]”).
295 Exh. 78, LinkedIn Profile for T. Laughren & PsychoPharm Consulting, at 1 (emphasis
added).
PART VI: FOREST USED THE FALSE ASSERTION THAT MD-18 WAS POSITIVE AND THE FDA’S APPROVAL FOR LEXAPRO TO NEGOTIATE REDUCED PENALTIES IN USAO CASE
When the USAO negotiated Forest’s criminal plea, civil settlement, and CIA, Forest was able
to use the “fact” that MD-18 was positive and that the FDA had approved, in March 2009, the
adolescent use of Lexapro. As it turns out, however, those two material facts, which clearly
limited the ability of the United States to fully prosecute Forest’s off-label promotion, were
based on outright fraud and deception. Under the terms of the criminal plea, the USAO retains
the right to reopen its prosecution if the plea agreement was based on or involved any
falsehoods.296 The information and evidence set forth in this memorandum strongly support the
reopening of the USAO’s prosecution of Forest, to hold Forest accountable for the fraud
perpetrated on the FDA, the USAO, physicians, parents, and the medical/scientific community.
296 Exh. 2, Plea Agreement at 6.




OVER $4 billion
in verdicts & Settlements
Our top priority is to devise customized legal strategies that are tailored to the unique legal needs of our clients, no matter how simple or complicated their situations, might be.
-
$265 Million Settlement Fatal Train Crash
In 2016, Wisner Baum attorney Timothy A. Loranger and six other attorneys in the Plaintiffs’ Management Committee were able to secure a $265 million settlement for victims of the 2015 Amtrak 188 derailment in Philadelphia, one of the largest in the U.S. for 2016.
-
$14 Million Settlement A Major US Plane Crash
Wisner Baum obtained a $14 million settlement for the death of a passenger in a major US plane crash.
-
$12 Million Settlement Helicopter Crash
Wisner Baum secured a $12 million settlement for a passenger who was injured in a helicopter crash.
-
$10 Million Settlement A Major Foreign Plane Crash
Wisner Baum obtained a $10 million settlement for the death of a passenger in a major foreign plane crash.
-
$2.0 Billion Verdict Personal Injury
In May of 2019, the jury in the case of Pilliod et al. v, Monsanto Company ordered the agrochemical giant to pay $2.055 billion in damages to the plaintiffs, Alva and Alberta Pilliod, a Bay Area couple in their 70s.

Our Case Results

-
$2.0 Billion Verdict Personal Injury
In May of 2019, the jury in the case of Pilliod et al. v, Monsanto Company ordered the agrochemical giant to pay $2.055 billion in damages to the plaintiffs, Alva and Alberta Pilliod, a Bay Area couple in their 70s.
-
$289.2 Million Verdict Personal Injury
On Aug. 10, 2018, a San Francisco jury ordered Monsanto to pay $39.25 million in compensatory damages and $250 million in punitive damages to Mr. Johnson, a former groundskeeper who alleged exposure to Monsanto’s herbicides caused him to develop terminal non-Hodgkin lymphoma.
-
$265 Million Settlement Fatal Train Crash
In 2016, Wisner Baum attorney Timothy A. Loranger and six other attorneys in the Plaintiffs’ Management Committee were able to secure a $265 million settlement for victims of the 2015 Amtrak 188 derailment in Philadelphia, one of the largest in the U.S. for 2016.
-
$105 Million Settlement Pharmaceutical Settlement
Wisner Baum obtained $105 million on behalf of multiple clients involved in a pharmaceutical negligence case.
-
$80 Million Verdict Personal Injury
Wisner Baum attorneys served on the trial team in the case of Hardeman v. Monsanto Company, which resulted in an $80 million jury verdict for the plaintiff, Edwin Hardeman.
-
$63 Million Settlement Paxil Pediatric Class Action
$63 million pediatric class action re false promotion of Paxil. Judge approves final terms of improved national pediatric paxil class action settlement – consumers get a better deal.
Client-Focused Representation
REVIEWS & TESTIMONIALS
We believe our track record speaks for itself. But you don’t have to take our word for it. See what our clients have to say about working with us.
-
"I Can’t Imagine a Better Law Firm"
Multiple lawyers recommended Wisner Baum to me and I have been consistently impressed with the quality of their work.
- Best Law Firms Survey -
"They Are About Changing the Systems..."
Wisner Baum are not only amazing attorneys but more importantly, they are activists. They are about changing the systems which got us into trouble in the first place. They understand their role in the process of making change.
- Kim Witczak -
"Top Legal Minds in the Country"
The Wisner Baum firm has some of the top legal minds in the country; they are driven, determined, trustworthy, ethical and passionate.
- From Best Lawyers® Best Law Firms


